Features and influencing mechanisms of gaseous elemental mercury over the equatorial Pacific and their differences with the Southern Ocean
2022-07-20WANGJianchengYUEFangeZHANHaicongKANGHuiXIEZhouqing
WANG Jiancheng, YUE Fange*, ZHAN Haicong, KANG Hui & XIE Zhouqing,2
Features and influencing mechanisms of gaseous elemental mercury over the equatorial Pacific and their differences with the Southern Ocean
WANG Jiancheng1, YUE Fange1*, ZHAN Haicong1, KANG Hui1& XIE Zhouqing1,2
1Institute of Polar Environment & Anhui Key Laboratory of Polar Environment and Global Change, Department of Environmental Science and Engineering, University of Science and Technology of China, Hefei 230026, China;2Center for Excellence in Urban Atmospheric Environment, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China
Due to the harmful impacts on the ecosystem and even human health, mercury (Hg) compounds in the environment deserve serious concern. Atmospheric mobilization and exchange at the air-sea interface are important processes in biogeochemical cycling of Hg. Relying on the 30th (2013/2014), 31st (2014/2015), and 33rd (2016/2017) Chinese National Antarctic Research Expedition aboard R/V, we found significant rising gaseous elemental mercury (GEM) concentrations over the equatorial Central Indo-Pacific region. Excluding the contribution of anthropogenic, volcanic and biomass burning emissions, the enhanced GEM in marine boundary layer was likely due to the combined actions of two driving factors drove by the Inter-Tropical Conversion Zone (ITCZ): (1) intense wet deposition of Hg, followed by subsequent rapid photoreduction and vast evasion from the surface sea; and (2) the regional low-level convergence of airflow that caused the mass accumulation of GEM in air. In addition, apparently higher GEM concentration level in the equatorial Central Indo-Pacific than in the Southern Ocean was observed in one cruise. Further investigation suggests that apart from the ITCZ corresponded mechanisms, the effects of spatial differences in anthropogenic emissions and more significant GEM oxidation in Antarctic sea should play roles in this phenomenon.
mercury, equatorial seas, Southern Ocean, rainfall,air-sea exchange, low-level convergence
1 Introduction
Mercury (Hg) is thought to global pollution with widespread health concern because of its highly toxic effect and bioaccumulation, especially methylated form (Ariya et al., 2015). In the atmosphere, Hg has three defined forms, which is gaseous elemental mercury (GEM, Hg0), gaseous oxidized mercury (GOM), and particulate bounded mercury (PBM). While it exists mainly (>90%) in the elemental phase (GEM) in the atmosphere which has a residence time about 0.5–2 a and could be long-range transported; reversely, GOM and PBM could be effectively removed from the air by dry and wet deposition with much shorter residence times (within few weeks) (Schroeder and Munthe, 1998; Lin and Pehkonen, 1999; Lindberg et al., 2007; Driscoll et al., 2013).
The air-sea exchange of Hg is complex, but critical for its global cycle (Mason and Sheu, 2002; Strode et al., 2007). Hg enters ocean mainly through wet deposition from atmosphere ( Sunderland and Mason, 2007; Holmes et al., 2010). After atmospheric Hg (especially reactive forms) deposited to the ocean, some of them could be transferred to methylated forms via abiotic and biotic reactions, and therefore bioaccumulate in the marine food chain,but most of them (>80%) would be back to marine boundary layer (MBL) via evasion by reduced to the dissolved gaseous Hg (DGM, mainly are Hg0) (Driscoll et al., 2013), so the surface ocean somehow looks like a temporary reservoir for Hg. Ocean evasion of Hg to the atmosphere was estimated approximately equal to anthropogenic emissions (Mason and Sheu, 2002; Pirrone and Mason, 2009). The elemental mercury (Hg0) which is subject to re-emission from the surface ocean is not only a main supply to Hg in the atmosphere, but also helps its long-range transport, this way to transport is also called as “grasshopper-jump” (Travnikov, 2012).
Previous studies in various ocean regions showed that the ocean had a strong tendency of (re)-emitting GEM to the troposphere as most sea surface waters were supersaturated of Hg0(Kim and Fitzgerald, 1986; Wängberg et al., 2001; Mason and Sheu, 2002; Gårdfeldt et al., 2003; Andersson et al., 2008, 2011; Fu et al., 2010; Ci et al., 2011, 2016; Kuss et al., 2011, 2015, 2018; Bagnato et al., 2013a; Soerensen et al., 2014; Wang et al., 2017). In a model study, Strode et al. (2007) indicated that the ocean emission contributed nearly half part of the atmospheric Hg in MBL, with the largest contribution from the tropics.
Actually, strong enrichment of DGM in surface water in equatorial ocean belts were reported in previous measurements (Kim and Fitzgerald, 1986; Kuss et al., 2011; Soerensen et al., 2014), and likely due to enhanced wet deposition by plentiful rainfall, associated with the Inter-Tropical Conversion Zone (ITCZ) (Kuss et al., 2011; Soerensen et al., 2014). Soerensen et al. (2014) also could simulate the peaks of DGM coincident with the ITCZ using the GEOS-Chem atmospheric transport model coupled to a global ocean model (MIT-gcm), although less than the observations.
Hg’s hemispheric gradient over the Atlantic and Pacific ocean has been reported in some studies, with higher concentrations in the North (Seiler et al., 1980; Slemr et al., 1981, 1985, 1995; Slemr and Langer, 1992; Temme et al., 2003; Soerensen et al., 2010, 2014; Xia et al., 2010; Müller et al., 2012), and usually interpreted as a direct evidence that the gaseous mercury could be transported globally (north with more anthropogenic emission to the south). But resent modeling study showed that the atmospheric Hg in the southern hemisphere is mostly originated from the oceanic evasion rather than transport from the northern hemisphere (Horowitz et al., 2017).
Despite the special role of the equatorial oceanic areas in the regional cycle of atmospheric and marine Hg, studies about the cycling of mercury in the tropical areas are still very scarce (Costa et al., 2012). As part of the 30th (2013/2014), 31st (2014/2015), and 33rd (2016/2017) Chinese National Antarctic Research Expeditions (CHINAREs) aboard R/V, here we report the continuous measurements of GEM in MBL cross the Central Indo-Pacific, along with some data of DGM. We further combined these data with the corresponding observations in the high-latitude Southern Ocean during the 31st CHINARE, to better understand the cycling of mercury in the tropical areas and its differences in the influencing mechanisms of atmospheric Hg with the Antarctic marine area.
2 Methods
2.1 Site locations
The sampling campaigns were conducted aboard R/V, which navigated across the Central Indo-Pacific, in the 30th (4 to 8, April 2014, leg 1), 31st (October 31, October to 10, November 2014, leg 2; 27, March to 6, April 2015, leg 3), and 33rd (29, March to 7, April 2017, leg 4) CHINAREs (Figure 1). In addition, the observation during the navigation across the high-latitude Southern Ocean (60°S–77.6°S; 70°E–180°E) in 31st CHINARE was also reported in this study.
2.2 Experimental methods
GEM, DGM, CO and O3were measured as described in our earlier studies (Yu et al., 2015; He et al., 2016; Wang et al., 2017) and is only briefly outlined here. GEM concentrations were continuously measured in all cruises, DGM concentrations were measured in the 33rd CHINARE, CO and O3mixing ratios were only measured in the 31st CHINARE. To the GEM, CO and O3,the air intakes for the monitoring instruments were located at the front of the icebreaker (about 15 m above the sea surface), opposite from the ship’s power system to minimize the impact of the smoke plume from the chimney. We further discarded any data point obtained when the icebreaker’s speed was ≤ 2 knot or when the wind was coming from the stern (relative wind direction to the heading ofbetween 120° and 240°). For measurement and analysis of DGM, seawater was manually collected from the sea surface (~ 0–1m depth) by a canvas bag at the front of the ship in leg 3, or from the clear seawater supply system in the ship that was continuously pumped below the ship’s hull (~ 4– 5 m depth) in leg 4. Samples were analyzed within 1 h by the purge and trap technique in a clean laboratory onboard the ship.
Meteorological, hydrologic and GPS data were obtained from the ship’s monitoring system; these included wind direction, wind speed (SPD), air pressure (P), relative humidity (RH), atmospheric temperature (AT), sea surface temperature (SST), salinity, ship direction and ship speed, all averaged over 5 min. Also, some data were acquired from NASA Earth Observations (NEO) (https://neo.sci. gsfc.nasa.gov), e.g., the SST (https://neo.sci.gsfc.nasa. gov/view.php?datasetId=MWOI_SST_M), sea surface salinity (https://neo.sci.gsfc.nasa.gov/view.php?datasetId= AQUARIUS_SSS_M), chlorophyll(chl) (https://neo. sci.gsfc.nasa.gov/view.php?datasetId=MY1DMM_CHLORA), active fires (https://neo.sci.gsfc.nasa.gov/view.php? datasetId=MOD14A1_M_FIRE), precipitation (https://disc. gsfc.nasa.gov/datasets/TRMM_3B42_Daily_V7/summary?keywords=%22TMPA%22%20and%20%22real-time%20daily%22). The volcanic activity and the SO2distributions were obtained from the Volcanic Hazards Project (https://so2.gsfc.nasa.gov), which were derived by Atmospheric Chemistry and Dynamics Laboratory, National Aeronautics and Space Administration (NASA). The HYSPLIT transport and dispersion model (Wang et al., 2009) from NOAA-ARL (Air Resources Laboratory)(http://www.arl.noaa.gov/ready/hysplit4.html) was used to identify the source of air masses from selected points. Maps of the BrO· vertical column were producted from Global Ozone Monitoring Experiment-2 (GOME-2) satellite data using the DOAS algorithm (http://www.iup.uni-bremen.de/ doas/scia_data_browser.htm). The cloud height data (which indicates the cloud top temperature) was derived from the match results between the brightness temperature data (https://www.ncei.noaa.gov/data/geostationary-ir-channel-brightness-temperature-gridsat-b1) and their corresponding vertical temperature profile (http://aura.gesdisc.eosdis.nasa. gov).

Figure 1 Tracks of the R/Vcruises across the Central Indo–Pacific in: a, the 30th CHINARE (April 4 to 8, 2014, leg 1); b, the 31st CHINARE (October 31 to November 10, 2014, leg 2); c, the 31st CHINARE (March 27 to April 6, 2015, leg 3); d, the 33rd CHINARE (March 29 to April 7, 2017, leg 4). The base map was generated by Ocean Data View 4.0.
3 Results and discussion
3.1 General characteristics and enhanced episodes of GEM over equatorial seas
The concentration of GEM observed in the equatorial Central Indo-Pacific in the 4 cruises displays a large spatial variation (Figure 2), with a concentration range from 0.36 ng·m−3to 31.94 ng·m−3, and an average of 3.80± 2.92 ng·m−3. This concentration level is apparently higher than that in some other oceanic areas (e.g. Chinese Yellow Sea (2.61 ± 0.50 ng·m−3); Mediterranean Sea (1.9 ± 1.0 ng·m−3); South China Sea (2.62 ± 1.13 ng·m−3); Arctic Ocean (1.7 ± 0.4 ng·m−3) and North Atlantic Ocean (1.7 ± 0.1 ng·m−3) (Sprovieri et al., 2003; Andersson et al., 2008, 2011; Fu et al., 2010; Ci et al., 2011). As we have filtered the GEM data to minimize the potential influence of ship emissions (please see the section 2.2), these concentration characteristics imply that some special mechanisms might drive the increase of GEM level in the equatorial Central Indo-Pacific.
As shown in Figure 2, pronounced GEMconcentration maximum was measured over equatorial seas (approximately defined by the latitudes 10° N and 10° S) in all cruises. In leg 1, the maximum of about 20 ng·m−3at 3°S–5°S, with nearly 10 times rising; In leg 2, the GEMmaximum of about 8 ng·m−3at 3°N–3°S, with nearly 4 times rising; In leg 3, the GEM maximum of about 30 ng·m−3at ~3° S, with nearly 5 times rising; In leg 4, the GEMmaximum of about 2.4 ng·m−3at ~8° S, with nearly 2 times rising. This is interesting because it breaks our previous knowledge about the gaseous Hg distribution between the two hemispheres. Most earlier measurements showed obvious Hg hemispheric gradient over the Atlantic and Pacific oceans (with higher concentrations in the north, and always lower to the south) (Slemr, 1996; Lamborg et al., 1999; Temme et al., 2003), also the model studies (Holmes et al., 2010; Soerensen, Sunderland, et al., 2010; Travnikov, 2012; Pacyna et al., 2016; Travnikov et al., 2017),but did not present the shifting in the equator, except the measurements by Fitzgerald et al. (1984) in the equatorial mid Pacific Ocean (TGM increased >0.5 ng·m−3).
3.2 Potential mechanisms driving the enhanced Hg in MBL
To identify these episodes of enhanced GEM observed over the equatorial seas, we investigated possible sources as below.

Figure 2 a1, a2, a3, a4 are the spatial distribution of GEM concentrations during the cruises in leg1, leg 2, leg3 and leg4, respectively;b1, The concentrations of GEM in the marine boundary layer, and the salinity in the surface seawater in leg 1; b2, The concentrations of GEM, carbon monoxide (CO) in the marine boundary layer, salinity (Sa), and Chlin the surface seawater in leg 2;b3, The concentrations of GEM, CO in the marine boundary layer, and the Sa, and Chlin the surface seawater in leg 3;b4, The concentrations of GEM in the marine boundary layer, and the dissolved gaseous mercury (DGM) in the surface seawater in leg 4. The grey segments identify the enhanced GEM values in b1, b2, b3, b4.
Plumes from the anthropogenic sources can extend into the MBL, as well as biomass burning (Temme et al., 2003; Fu et al., 2010; Xia et al., 2010). Anthropogenicsources of Hg from the terrestrial boundary layer could infect the marine boundary layer, especially near the coast. South-East Asia has large coastal population densities, and the active human activities are believed to be important Hg sources (Pacyna et al., 2010). However, Sørensen (2011) indicated that the anthropogenic Hg would dilute rapidly when the plumes left not far away to the open ocean. Biomass burning plumes usually contain vast Hg, and could be transported long away(Friedli et al., 2009; Xia et al., 2010; Webster et al., 2016; Fraser et al., 2018). However, the backward air trajectories did not show the transport of anthropogenic sources in all the legs, combined the location of big cities and fires (scanned from the web:https://firms. modaps.eosdis.nasa.gov/firemap/). On the contrary, the air masses when the GEM showed higher values were all from the open seas (Figure 3).
GEM/CO relationship can be used to identity alternative sources since the known emission fingerprints. GEM/CO ratios were reported as 0.0005–0.002 ng·m−3·ppbv−1for biomass burning (Brunke et al., 2001; Ebinghaus et al., 2007; Weisspenzias et al., 2007; Friedli et al., 2009; Sørensen, 2011), and 0.0013–0.006 ng·m−3·ppbv−1for anthropogenic pollutions, respectively (Weisspenzias et al., 2007; Obrist et al., 2008; Sørensen, 2011). In this study, observed GEM/CO ratio ranges from 0.0072 to 0.38 ng·m−3·ppbv−1. These high GEM/CO ratios did not support the substantial contribution of anthropogenic or biomass burning sources in our study.

Figure 3 168-hour air mass backward trajectory when the GEM presented higher values in all cruises. a, 1:00, 5 Apr, 2014, leg 1; b, 9:00, 4 Nov, 2014, leg 2; c, 15:00, 6 Nov, 2014, leg 2; d, 8:00, 2 Apr, 2015, leg 3; e, 22:00, 31 Mar, 2017, leg 4.
Volcanic sources sometime also might be involved in elevated atmospheric Hg (Nriagu and Becker, 2003; Pyle and Mather, 2003; Bagnato et al., 2013b; Bagnato et al., 2015). Enhanced atmospheric Hg from volcanic emissions had been observed in MBL in previous measurements (Xia et al., 2010; Yu et al., 2015). Indeed, many active volcanoes are located around the Central Indo-Pacific (e.g. in Philippines, Indonesia, and New Guinea). However, the backward air trajectories and the combined SO2distribution (scanned from the web: https://so2.gsfc.nasa.gov) still did not show any of these plumes was mixed with volcanic sources in all legs (Figure A1).
In early studies, Fitzgerald et al. (1984)and Kim and Fitzgerald (1986) reported enhanced seawater DGM and associated evasion in equatorial Mid Pacific Ocean and attributed it to higher biological reduction of enhanced reactive Hg in productive upwelling regions, but the anti-correlated correlation between the GEM and seawater salinity in this study indicated that the Hg was not mainly supplied by the upwelling as the deep seawater has higher salinity. The GEM series also had no correspondence with the corresponding Chl-in leg 2 and leg 3 (Figure 2), so the biological productivities, which would cause the biotic reduction of aquatic Hg species, did not seem to be a major contributor (Soerensen et al., 2014).
3.2.1 The effect of the Hg wet-deposition and evasion on the enhancement of GEM
Excluding those possibilities, supplement from the ocean evasion was then considered. The measurements of study in low latitude Atlantic and Pacific Oceans showed the highest DGM concentrations in the surface sea (Kuss et al., 2011), especially in the ITCZ. Enhanced DGM concentrations in surface water in equatorial seas was also found in our measurement in leg 4, and correlated with the enhanced GEM in marine boundary layer (Figure 2). Monthly average DGM concentrations by MIT-gcm Model were always significantly elevated in equatorial seas, and mostly correlated with our measured GEM (Figure 4). The vast evasion of Hg from the equatorial seas to replenish the atmosphere seems plausible.

Figure 4 GEM concentrations in MBL, and DGM concentrations (by GEOS-Chem Model in monthly average) in surface seawater in leg 1 (a), leg 2 (b), leg 3 (c), leg 4 (d).
Atmospheric wet and dry deposition is the main source of Hg to the ocean. Wet deposition could bring vast mercury as the existence of higher total Hg concentrations in rainfall (about 20−50 times than seawater) (Soerensen et al., 2014). Horowitz et al. (2017) indicated that the 80% global wet deposition of Hg took place over the marine area, and most were in tropics. Kuss et al. (2011) reported the enhanced total Hg concentrations in surface seawater in the equatorial seas in Atlantic, and attributed it to the increased supply from the rainfall. After deposition, these divalent Hg (HgII) are likely fast photoreduced to element form (Hg0) due to intense solar radiation (Kuss et al., 2011; Ci et al., 2016).
The equatorial Central Indo-Pacific has the most bountiful rainfall in the world. Rainfall in the equatorial areas is much higher than adjacent areas, so the wet deposition of Hg would be significant. The enhanced GEM in MBL was anti-correlated with the seawater salinity in leg 1, leg 2, and leg 3 (Figure 2, light grey frame), which implied the role of precipitation in diluting the seawater. Actually, Horowitz et al. (2017)simulated that higher wet deposition of Hg would occur in tropical Central Indo-Pacific Ocean due to elevated precipitation using GEOS-Chem model. On account of the lack of the corresponding mercury wet deposition observation during this cruise, here we just investigate the precipitation characteristics during our cruise and generally estimate their impact on GEM concentration. In view of the time period of the reduction and re-emission of the HgIIbrought by wet deposition and the storage time of GEM in air, the 7-day total precipitation data of leg 1, leg 2 and leg 3 was displayed here (Figure 5) (due to the obviously lower concentration of GEM, we haven’t paid attention to leg 4 here). It showed that the equatorial Central Indo-Pacific region had undergone various degrees of heavy precipitations, which could even exceed 200 mm during our cruise, and most concentration peak of GEM were accompanied well with apparently elevated precipitation, especially in leg 1 and leg 2. The DGM concentrations simulated by MIT-gcm in leg 1 and leg 2 were also in significant correlation with our observed salinity (Figure A2). As the heavy precipitation would dilute sea water and cause the reduced salinity, these were to some point consistent with our assumption.
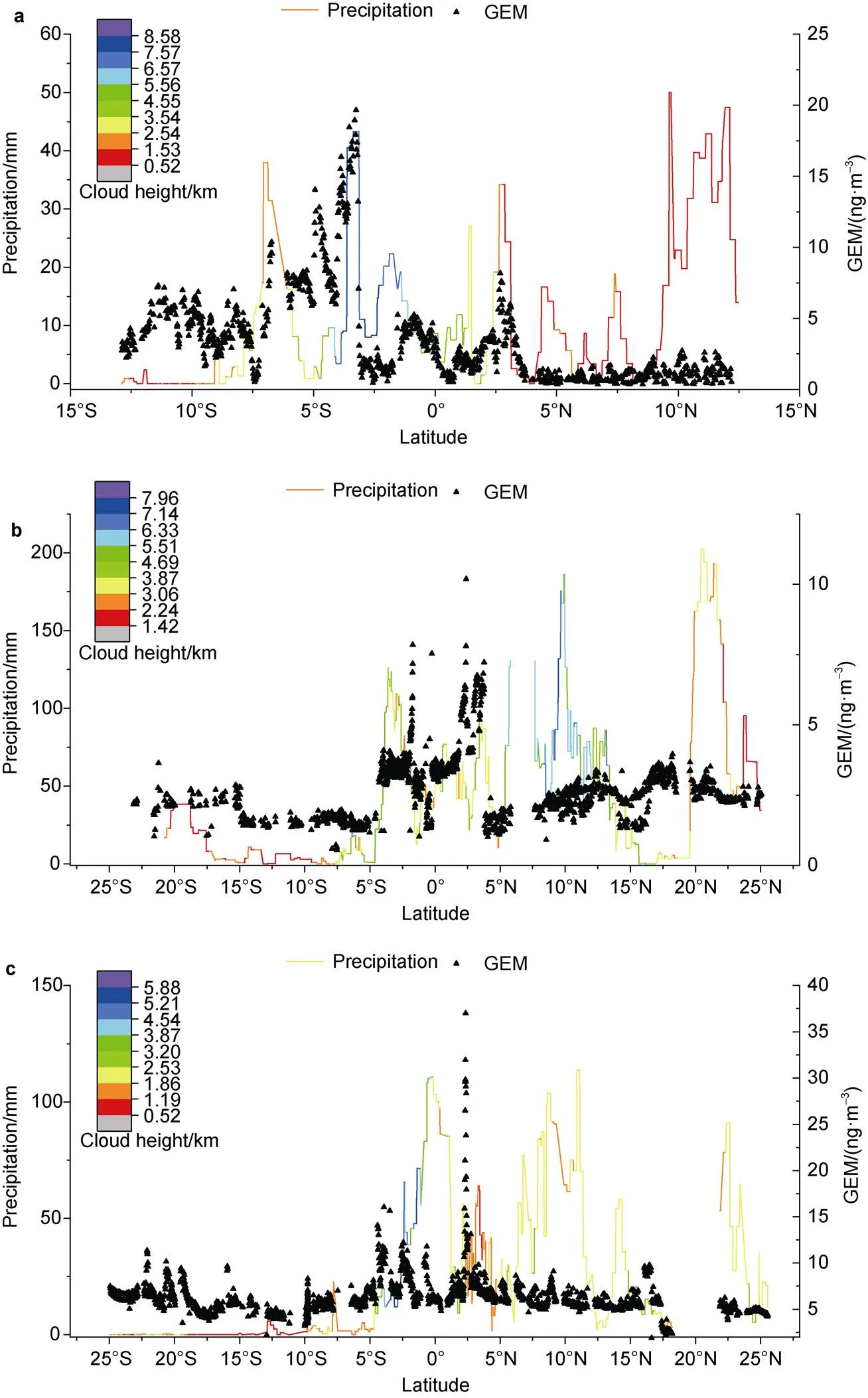
Figure 5 The latitude series of GEM concentrations (the black scatter plot) accompanied with their corresponding 7-days total precipitation data (the line plot) in leg1 (a), leg2 (b) and leg3 (c); the color mapping of each line plot was the corresponding 7-days average cloud height data.
In addition to high precipitation, frequent deep convection in ITCZ would further intensify Hg wet deposition (Holmes et al., 2016). As the concentration of HgIIcan be about 2 orders of magnitude higher in the upper troposphere than at the surface due to higher bromine mass and lower temperature, which would promote the formation of HgII, and then would be easily scavenged by rainfall (Holmes et al., 2010; Lyman and Jaffe, 2012). In a model study, Soerensen et al. (2014) indicated that the MIT-gcm model would underestimate the magnitude of the seawater DGM concentrations compared to their observed peaks in the ITCZ of both the Atlantic and Pacific oceans, and attributed it to insufficient consideration about the deep convective scavenging of Hg from the upper troposphere. Zhang et al. (2019) also found that the regional cloud height could be up to more than 8 km when the observed dissolved gaseous mercury peaked. Here the 7-days average cloud height data were added to GEM and precipitation series. The GEM maximum and precipitation peak matched well with the highest average cloud height which could rose up to more than 8 km in leg 1, indicating the potential importance of deep convective precipitation in the rising concentrations of GEM in this cruise, while they were not in good correspondence in leg 2 and leg 3. It may be due to the contingency of deep convective precipitation, as the 7-days averaging treatment of cloud height data may pulled down the temporary deep convection meanwhile (e.g. the cloud height could grew up to 7–10 km before the GEM peak occurred in leg 2 and even 12 km in leg 3). Based on the sensitive analysis in model research, Zhang et al. (2019) suggested that the concentrations of DGM along the ITCZ are sensitive to the deep convection strength above 600 hpa and indicate the important role of active deep convection in the elevated seawater Hg concentrations in this region. On account of the sparse researches of Hg wet deposition in equatorial west Pacific Ocean, the role of deep convective precipitation of Hg needs further investigation.
The ITCZ is not only characterized by intense rainfall, but also a region of low-level mass convergence (Byrne and Schneider, 2016). During the exploring process of deep convective precipitation, we occasionally found that there were good correspondences between the GEM concentration series and its corresponding daily cloud height series (one day before our cruise) in leg 1, leg 2 and leg3 (Figure 6). It may implied that apart from intense precipitation, another meteorological process—regional convergence, may also contribute to the enhanced GEM.
3.2.2 The effect of the low-level convergence on the enhancement of GEM
In general, cloud is mainly generated through the adiabatic expansion and cooling of the saturated moist air mass during its rising process, so sufficient water vapor and its updraft are two necessary points for the formation of clouds. The convective updraft is usually correlated with the low-level convergence (Raymond et al., 2003), hence high cloud height may indicate the strong low-level convergence. Ascending motion of stronger convergent convection would not only take low-level moisture to higher altitude to form cloud, but also apparently gather the atmospheric mercury through the low-level compensated air-mass (Ekman-pump), which would cause the rise of the GEM concentration (the 7-days average cloud height data also have similar variation trend with the GEM series, suggests that this regional convergence may be a long-term continuous process). We then compared the corresponding daily sea-level pressure data with cloud height, and it presented that higher cloud height tend to correspond to lower sea-level pressure (Figure 6), which further supported our speculation.
Previous studies suggested the rainfall would bring vast Hg to the equatorial seas (ITCZ) and cause the enhanced DGM, but most did not show the enhanced GEM in the MBL (e.g. Kuss et al. (2011) and Soerensen et al. (2014)). Besides, Wang et al. (2014) observed no sustained high GEM in the equatorial eastern Pacific Ocean in one year-round measurements of species Hg on Galápagos Islands, and indicated no persistently enhanced mercury evasion from the upwelling sea. But our results showed the obvious variance of GEM in the MBL over the equatorial seas in Central Indo-Pacific, also the measurements by Fitzgerald et al. (1984) in the equatorial mid Pacific Ocean (TGM increased >0.5 ng·m−3). We speculated the reason might be the spatial and temporal variance of rainfall and regional convergence. It should be noted that the rainfall and regional convergence are occasional events, and they have big spatial variance in the equator. Driven by the Walker Circulation(Wikipedia, 2017), the western side of the equatorial Pacific is wetter due to more frequent rainfall caused by moist convective updraft, while the eastern side is relatively dryer, so the extent and frequency of wet deposition and convergence- driven gathering of mercury in equatorial west Pacific Ocean would be larger than that in other equatorial seas in general, just as the results of a model study by Costa et al. (2012). Horowitz et al. (2017) also simulated that the biggest wet deposition of Hg would occur in the Central Indo-Pacific. To some extent, this could explain the more remarkable enhanced GEM in MBL over this study area. As an evidence, Mason et al. (2017) reported enhanced GEM concentrations in MBL and concurrent higher DGM concentrations in surface water in west low latitude Pacific Ocean than that in the similar latitude in east regions, and ascribed it to the westward increase of wet deposition of Hg by the higher precipitation and subsequent the greater ocean evasion in there.
Moreover, the region we observed the enhanced GEM during this cruises were mainly located in the equatorial calm zone (5°S–5°S). It is characterized by small horizontal pressure gradients, which will be not conducive to the horizontal diffusion of pollutants. ITCZ itself is a vast convergence zone. As the zone with the lowest pressure between the two subtropical high pressure areas, ITCZ may have a general tendency of convergent mercury accumulation in the lower atmosphere. Under this condition, regional low-level convergence, heavy convective precipitation, conspicuous photo reduction and high temperature-driven re-emission would jointly lead to obvious increasing of GEM. Due to the sparse data of Hg wet deposition, and other probable undetected mechanisms in this region. The significantly enhanced GEM in the equatorial Central Indo-Pacific area need further explorations in the future.
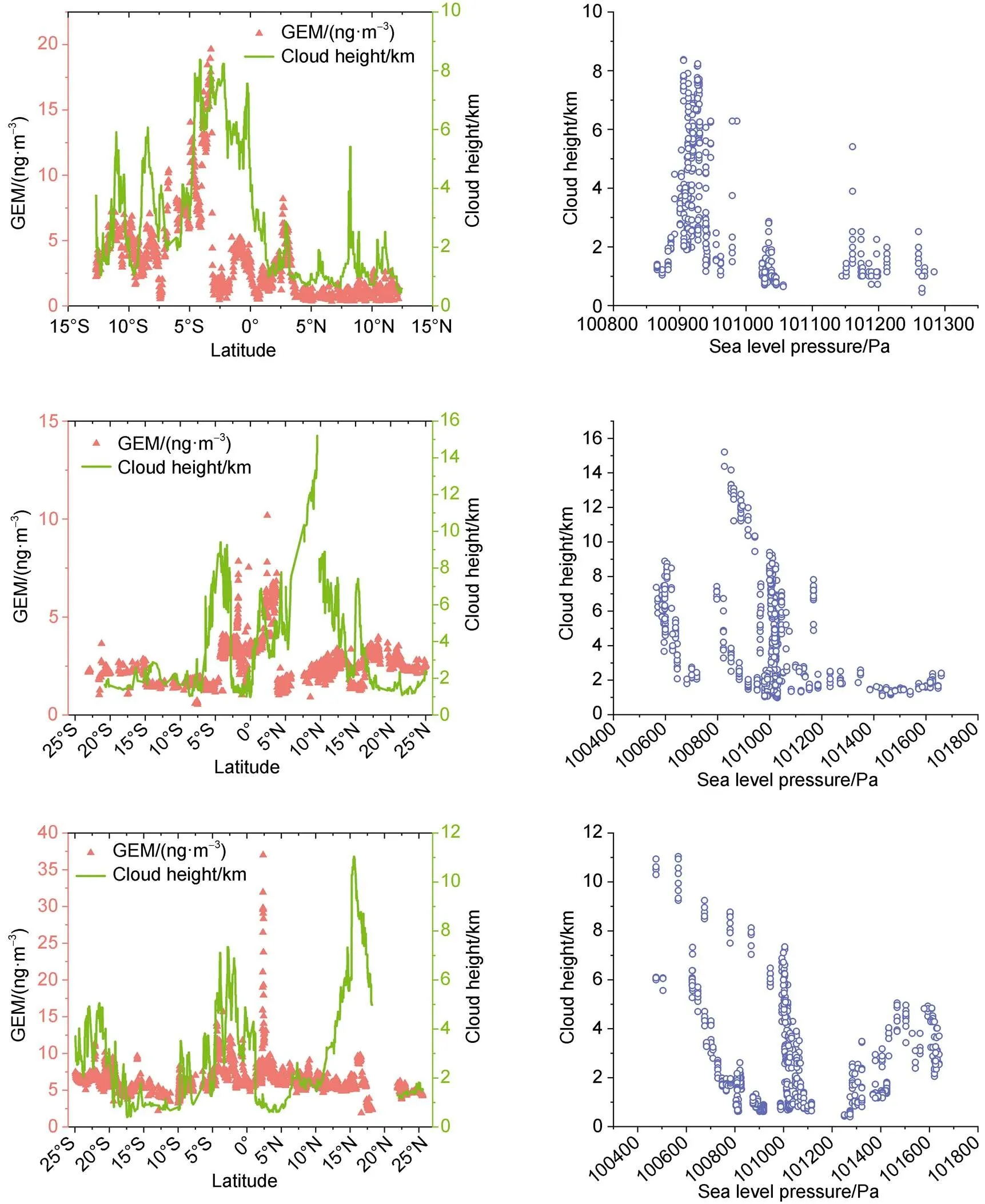
Figure 6 The latitude series of GEM concentrations accompanied with its corresponding daily-averaged cloud height (left) and the relationship between daily-averaged cloud height and daily sea level pressure (right) in leg 1 (a), leg 2 (b) and leg 3 (c).
3.3 Comparison between the equatorial Central Indo-Pacific area and the Southern Ocean
As GEM was also continuously observed in the high-latitude Southern Ocean (south of 60°S) during the 31st CHINARE, a comparison of the observed GEM between the equatorial Central Indo-Pacific area and the Southern Ocean in this cruise is conducted in this study, aimed at investigating the differences in the influencing mechanisms of GEM in these two oceanic areas.
The observed GEM in the Southern Ocean ranges from 0.39 to 1.92 ng·m−3, with an average of 0.93±0.19 ng·m−3. This concentration level is similar to the results observed in the Antarctic coastal or oceanic area that reported in previous studies (e.g. the Ross Sea (0.96 ± 0.21 ng·m−3); and the Antarctic Terra Nova Bay (0.9±0.3 ng·m−3) (Sprovieri et al., 2002; Yue et al., 2021), while is obviously lower than that in the equatorial Central Indo-Pacific area (Figure 7). Apart from the intense wet deposition and subsequent vast evasion of Hg from the surface sea, and the regional low-level convergence of airflow that caused the mass accumulation of Hg in air, which were discussed before, the effects of the differences in anthropogenic emissions and more significant GEM oxidation in Antarctic sea should play roles in it. As displayed in Figure 7, the general concentration level of observed CO was apparently higher in the equatorial Central Indo-Pacific area (90.32 ± 34.59 ppbv) than in the Southern Ocean (48.47±11.67 ppbv), indicating more intensive human activities in the low-middle latitude area would effectively increase the background content of atmospheric Hg. In addition, due to the special photochemical mechanisms among the interface of snowpack and sea-ice, Antarctic area has high oxidative capacity of atmospheric Hg. Significant oxidation of GEM in the Antarctic area during summertime has been reported in several previous studies, which was also found in this cruise (Angot et al., 2016; Yue et al., 2021). For instance, significant depletion of GEM concentrations have be observed on 27 and 29, December 2014 in the Antarctic Ross Sea, with GEM concentrations dropping to nearly 0.6 and 0.4 ng·m−3respectively. Meanwhile, the corresponding observed O3concentrations show similar variation trends with GEM (the orange shaded area in Figure 8). This would highlight the role of halogen radicals in GEM oxidation in the Antarctic area, as the reactive halogen radicals (e.g. Br·, BrO·) formed photochemically upon the interface of snowpack and sea-ice can oxidize both GEM and O3in the air, resulting in their synchronous concentration depletions (Lindberg et al., 2002; Steffen et al., 2013). This viewpoint is also supported by the GOME-2 satellite maps, which displayed high BrO· column concentrations near our study sites (red circle parts in Figure 8). Therefore, more significant GEM oxidation in the Southern Ocean will also contribute to the lower GEM concentration level in the Southern Ocean than that in the equatorial Central Indo-Pacific area.
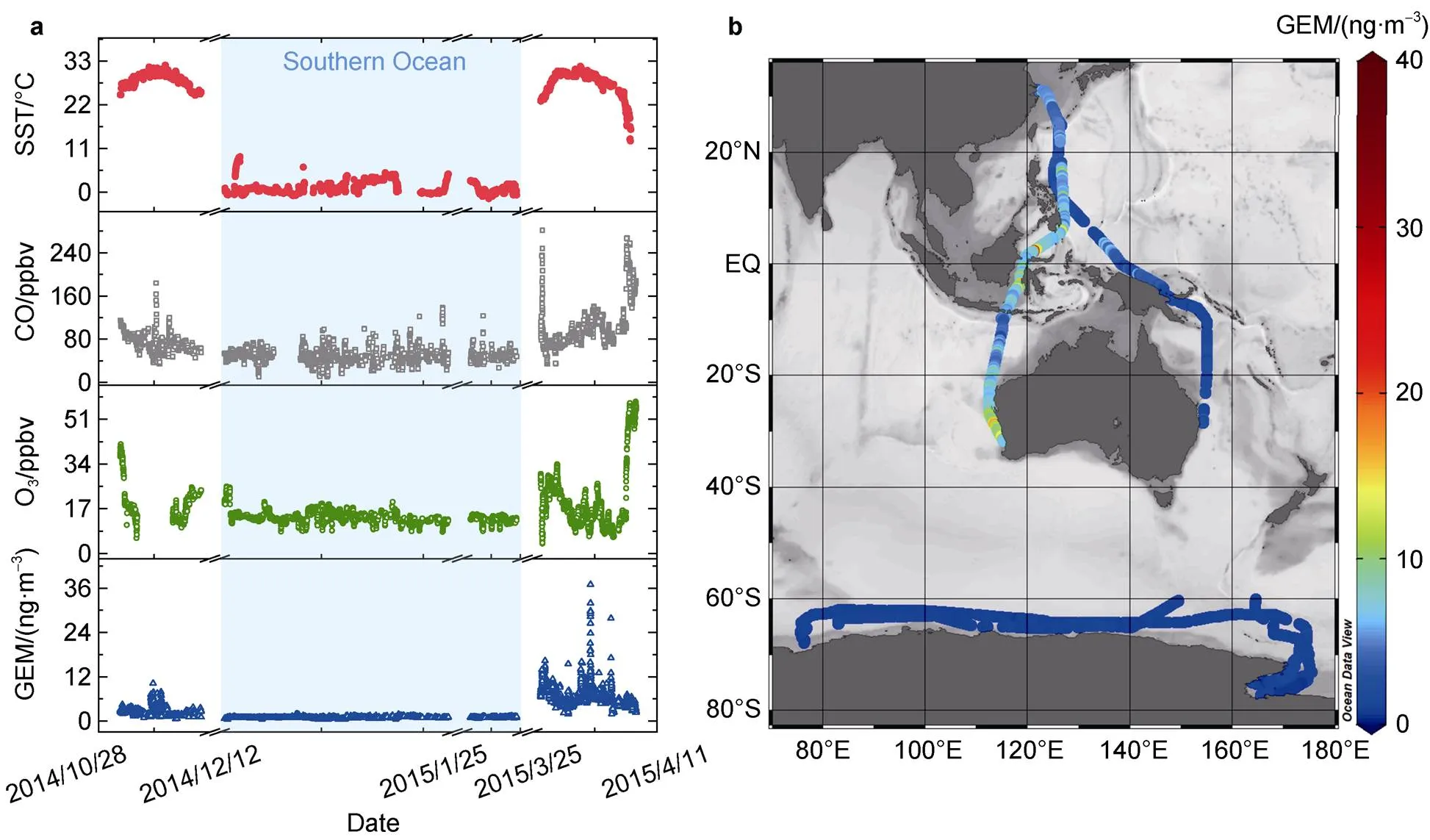
Figure 7 a, Time series for Gaseous Elemental Mercury (GEM), O3, CO and Sea Surface Temperature (SST) in the equatorial Central Indo-Pacific area and the Southern Ocean, which is marked with blue shading; b, The corresponding spatial distribution of GEM in the equatorial Central Indo-Pacific area and the Southern Ocean during the 34th CHINARE.

Figure 8 a, Relationship between GEM and O3during 26–29 December 2014 in the Southern Ocean; b, The GOME-2 maps of BrO· vertical column density distribution on 27 and 29 December 2014.
4 Conclusions and implications
We found clearly shifting GEM concentrations over the equatorial seas in Central Indo-Pacific, which would be attributed to the elevated marine Hg evasion driven by the vast Hg wet deposition, and regional low-level convergence of airflow in this area. The Pacific walker circulation at western-pacific equatorial areas favors the formation of heavy convective rainfall. If accompanied with deep convection, the Hg input will become more significant. As the strong updraft will reach the upper troposphere where the GOM species are obviously enhanced and easily scavenged by the rainfall. Moreover, the frequent low-level convergence in this region would facilitate the gathering of atmospheric mercury, and it may be exacerbated by the accumulative effect of equatorial calm zone, which is characterized by small horizontal pressure gradients and is distributed with many potential mercury emission sources in Southeast Asia (Yue et al., 2022). The comparison of GEM observations showed obviously higher GEM concentration level in the equatorial Central Indo-Pacific than in the Southern Ocean, which will be to some extent attributed to the effects of spatial differences of anthropogenic emissions and more significant GEM oxidation in Antarctic oceanic area.
Acknowledgments This work was financially supported by National Polar Special Program “Impact and Response of Antarctic Seas to Climate Change” (Grant no. 01-01-02E), and was supported by the National Natural Science Foundation of China (Grant no. 41941014). We thank Chinese Arctic and Antarctic Administration and Polar Research Institute of China for fieldwork support. We thank Yanxu Zhang at Nanjing University for helpful discussion and improving English expression. The authors acknowledge the NOAA Air Resources Laboratory (ARL) for developing the HYSPLIT transport and dispersion model available online (https://www.ready.noaa.gov/ HYSPLIT.php). We appreciate two anonymous reviewers, and Guest Editor Dr. Jianmin Pang for their constructive comments that have further improved the manuscript.
Note Wang Jiancheng and Yue Fangehave contributed to this work equally.
Andersson M E, Sommar J, Gårdfeldt K, et al. 2011. Air-sea exchange of volatile mercury in the North Atlantic Ocean. Mar Chem, 125(1-4): 1-7, doi:10.1016/j.marchem.2011.01.005.
Andersson M E, Sommar J, Gårdfeldt K, et al. 2008. Enhanced concentrations of dissolved gaseous mercury in the surface waters of the Arctic Ocean. Mar Chem, 110(3-4): 190-194, doi:10.1016/j. marchem.2008.04.002.
Angot H, Magand O, Helmig D, et al. 2016. New insights into the atmospheric mercury cycling in central Antarctica and implications on a continental scale. Atmos Chem Phys, 16(13): 8249-8264, doi:10.5194/acp-16-8249-2016.
Ariya P A, Amyot M, Dastoor A, et al. 2015. Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: a review and future directions. Chem Rev, 115(10): 3760-3802, doi:10.1021/cr500667e.
Bagnato E, Sproveri M, Barra M, et al. 2013. The sea-air exchange of mercury (Hg) in the marine boundary layer of the Augusta Basin (southern Italy): concentrations and evasion flux. Chemosphere, 93(9): 2024-2032, doi:10.1016/j.chemosphere.2013.07.025.
Bagnato E, Tamburello G, Aiuppa A, et al. 2013. Mercury emissions from soils and fumaroles of Nea Kameni volcanic centre, Santorini (Greece). Geochem J, 47(4): 437-450, doi:10.2343/geochemj.2.0263.
Bagnato E, Tamburello G, Avard G, et al. 2015. Mercury fluxes from volcanic and geothermal sources: an update. Geol Soc Lond Special Publ, 410(1): 263-285, doi:10.1144/sp410.2.
Brunke E G, Labuschagne C, Slemr F. 2001. Gaseous mercury emissions from a fire in the Cape Peninsula, South Africa, during January 2000. Geophys Res Lett, 28(8): 1483-1486, doi:10.1029/2000GL0 12193.
Byrne M P, Schneider T. 2016. Narrowing of the ITCZ in a warming climate: physical mechanisms. Geophys Res Lett, 43(21): 11350- 11357, doi:10.1002/2016GL070396.
Ci Z, Zhang X, Wang Z. 2016. Air-sea exchange of gaseous mercury in the tropical coast (Luhuitou fringing reef) of the South China Sea, the Hainan Island, China. Environ Sci Pollut Res Int, 23(11): 11323- 11329, doi:10.1007/s11356-016-6346-5.
Ci Z, Zhang X, Yin Y, et al. 2016. Mercury redox chemistry in waters of the eastern Asian seas: from polluted coast to clean open ocean. Environ Sci Technol, 50(5): 2371-2380, doi:10.1021/acs.est.5b05372.
Ci Z J, Zhang X S, Wang Z W, et al. 2011. Distribution and air-sea exchange of mercury (Hg) in the Yellow Sea. Atmos Chem Phys, 11(6): 2881-2892, doi:10.5194/acp-11-2881-2011.
Costa M F, Landing W M, Kehrig H A, et al. 2012. Mercury in tropical and subtropical coastal environments. Environ Res, 119: 88-100, doi:10.1016/j.envres.2012.07.008.
Driscoll C T, Mason R P, Chan H M, et al. 2013. Mercury as a global pollutant: sources, pathways, and effects. Environ Sci Technol, 47(10): 4967-4983, doi:10.1021/es305071v.
Ebinghaus R, Slemr F, Brenninkmeijer C A M, et al. 2007. Emissions of gaseous mercury from biomass burning in South America in 2005 observed during CARIBIC flights. Geophys Res Lett, 34(8): L08813, doi:10.1029/2006GL028866.
Fitzgerald W F, Gill G A, Kim J P. 1984. An equatorial Pacific Ocean source of atmospheric mercury. Science, 224(4649): 597-599, doi:10.1126/science.224.4649.597.
Fraser A, Dastoor A, Ryjkov A. 2018. How important is biomass burning in Canada to mercury contamination? Atmos Chem Phys, 18(10): 7263-7286, doi:10.5194/acp-18-7263-2018.
Friedli H, Arellano A F, Cinnirella S, et al. 2009. Initial estimates of mercury emissions to the atmosphere from global biomass burning. Environ Sci Technol, 43(10): 3507-3513, doi:10.1021/es802703g.
Fu X, Feng X, Zhang G, et al. 2010. Mercury in the marine boundary layer and seawater of the South China Sea: Concentrations, sea/air flux, and implication for land outflow. J Geophys Res Atmos, 115(D6): D06303, doi:10.1029/2009JD012958.
Gårdfeldt K, Sommar J, Ferrara R, et al. 2003. Evasion of mercury from coastal and open waters of the Atlantic Ocean and the Mediterranean Sea. Atmos Environ, 37: 73-84, doi:10.1016/S1352- 2310(03)00238-3.
He P Z, Bian L G, Zheng X D, et al. 2016. Observation of surface ozone in the marine boundary layer along a cruise through the Arctic Ocean: from offshore to remote. Atmos Res, 169: 191-198, doi:10.1016/j. atmosres.2015.10.009.
Holmes C D, Jacob D J, Corbitt E S, et al. 2010. Global atmospheric model for mercury including oxidation by bromine atoms. Atmos Chem Phys, 10(24): 12037-12057, doi:10.5194/acp-10-12037-2010.
Holmes C D, Jacob D J, Corbitt E S, et al. 2010. Global atmospheric model for mercury including oxidation by bromine atoms. Atmos Chem Phys, 10(24): 12037-12057, doi:10.5194/acp-10-12037-2010.
Holmes C D, Krishnamurthy N P, Caffrey J M, et al. 2016. Thunderstorms increase mercury wet deposition. Environ Sci Technol, 50(17): 9343-9350, doi:10.1021/acs.est.6b02586.
Horowitz H M, Jacob D J, Zhang Y, et al. 2017. A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget. Atmos Chem Phys, 17(10): 6353-6371, doi:10.5194/acp-17-6353-2017.
Kim J P, Fitzgerald W F. 1986. Sea-air partitioning of mercury in the equatorial Pacific Ocean. Science, 231(4742): 1131-1133, doi:10. 1126/science.231.4742.1131.
Kuss J, Krüger S, Ruickoldt J, et al. 2018. High-resolution measurements of elemental mercury in surface water for an improved quantitative understanding of the Baltic Sea as a source of atmospheric mercury. Atmos Chem Phys, 18(6): 4361-4376, doi:10.5194/acp-18-4361- 2018.
Kuss J, Wasmund N, Nausch G, et al. 2015. Mercury emission by the Baltic Sea: a consequence of cyanobacterial activity, photochemistry, and low-light mercury transformation. Environ Sci Technol, 49(19): 11449-11457, doi:10.1021/acs.est.5b02204.
Kuss J, Zülicke C, Pohl C, et al. 2011. Atlantic mercury emission determined from continuous analysis of the elemental mercury sea-air concentration difference within transects between 50°N and 50°S. Glob Biogeochem Cycles, 25(3): GB3021, doi:10.1029/ 2010GB 003998.
Lamborg C H, Rolfhus K R, Fitzgerald W F, et al. 1999. The atmospheric cycling and air-sea exchange of mercury species in the South and equatorial Atlantic Ocean. Deep Sea Res Part II Top Stud Oceanogr, 46(5): 957-977, doi:10.1016/S0967-0645(99)00011-9.
Lin C J, Pehkonen S O. 1999. The chemistry of atmospheric mercury: a review. Atmos Environ, 33(13): 2067-2079, doi:10.1016/S1352- 2310(98)00387-2.
Lindberg S, Bullock R, Ebinghaus R, et al. 2007. A synthesis of progress and uncertainties in attributing the sources of mercury in deposition. AMBIO A J Hum Environ, 36(1): 19-33, doi:10.1579/0044-7447 (2007)36[19: asopau]2.0.co;2
Lindberg S E, Brooks S, Lin C J, et al. 2002. Dynamic oxidation of gaseous mercury in the Arctic troposphere at polar sunrise. Environ Sci Technol, 36(6): 1245-1256, doi:10.1021/es0111941.
Lyman S N, Jaffe D A. 2012. Formation and fate of oxidized mercury in the upper troposphere and lower stratosphere. Nat Geosci, 5(2): 114-117, doi:10.1038/ngeo1353.
Mason R P, Hammerschmidt C R, Lamborg C H, et al. 2017. The air-sea exchange of mercury in the low latitude Pacific and Atlantic Oceans. Deep Sea Res Part I Oceanogr Res Pap, 122: 17-28, doi:10.1016/j. dsr.2017.01.015.
Mason R P, Sheu G R. 2002. Role of the ocean in the global mercury cycle. Glob Biogeochem Cycles, 16(4): 40-1, doi:10.1029/2001GB001440.
Müller D, Wip D, Warneke T, et al. 2012. Sources of atmospheric mercury in the tropics: continuous observations at a coastal site in Suriname. Atmos Chem Phys, 12(16): 7391-7397, doi:10.5194/ acp-12-7391- 2012.
Nriagu J, Becker C. 2003. Volcanic emissions of mercury to the atmosphere: global and regional inventories. Sci Total Environ, 304(1-3): 3-12, doi:10.1016/S0048-9697(02)00552-1.
Obrist D, Hallar A G, McCubbin I, et al. 2008. Atmospheric mercury concentrations at Storm Peak Laboratory in the Rocky Mountains: evidence for long-range transport from Asia, boundary layer contributions, and plant mercury uptake. Atmos Environ, 42(33): 7579-7589, doi:10.1016/j.atmosenv.2008.06.051.
Pacyna E G, Pacyna J M, Sundseth K, et al. 2010. Global emission of mercury to the atmosphere from anthropogenic sources in 2005 and projections to 2020. Atmos Environ, 44(20): 2487-2499, doi:10.1016/j.atmosenv.2009.06.009.
Pacyna J M, Travnikov O, de Simone F, et al. 2016. Current and future levels of mercury atmospheric pollution on a global scale. Atmos Chem Phys, 16(19): 12495-12511, doi:10.5194/acp-16-12495-2016.
Pirrone N and Mason R. 2009. Mercury fate and transport in the global atmosphere: emissions, measurements and models. Springer, Boston, MA, doi: 10.1007/978-0-387-93958-2.
Pyle D M, Mather T A. 2003. The importance of volcanic emissions for the global atmospheric mercury cycle. Atmos Environ, 37(36): 5115-5124, doi:10.1016/j.atmosenv.2003.07.011.
Raymond D J, Raga G B, Bretherton C S, et al. 2003. Convective forcing in the intertropical convergence zone of the eastern Pacific. J Atmos Sci, 60(17): 2064-2082, doi:10.1175/1520-0469(2003)060<2064: cfitic>2.0.co;2.
Schroeder W H, Munthe J. 1998. Atmospheric mercury—an overview. Atmos Environ, 32(5): 809-822, doi:10.1016/S1352-2310(97)00293-8.
Seiler W, Eberling C, Slemr F. 1980. Global distribution of gaseous mercury in the troposphere. Pure Appl Geophys, 118(2): 964-974, doi:10.1007/BF01593043.
Slemr F. 1996. Trends in atmospheric mercury concentrations over the Atlantic Ocean and at the Wank Summit, and the resulting constraints on the budget of atmospheric mercury//Baeyens W, Ebinghaus R, Vasiliev O (eds). Global and regional mercury cycles: sources, fluxes and mass balances. NATO ASI Series (Series 2: Environment), vol 21. Springer, 33-84, doi:10.1007/978-94-009-1780-4_2.
Slemr F, Junkermann W, Schmidt R W H, et al. 1995. Indication of change in global and regional trends of atmospheric Mercury concentrations. Geophys Res Lett, 22(16): 2143-2146, doi:10.1029/95GL01790.
Slemr F, Langer E. 1992. Increase in global atmospheric concentrations of mercury inferred from measurements over the Atlantic Ocean. Nature, 355(6359): 434-437, doi:10.1038/355434a0.
Slemr F, Schuster G, Seiler W. 1985. Distribution, speciation, and budget of atmospheric mercury. J Atmos Chem, 3(4): 407-434, doi:10.1007/BF00053870.
Slemr F, Seiler W, Schuster G. 1981. Latitudinal distribution of mercury over the Atlantic Ocean. J Geophys Res, 86(C2): 1159, doi:10.1029/ jc086ic02p01159.
Soerensen A L, Mason R P, Balcom P H, et al. 2014. Elemental mercury concentrations and fluxes in the tropical atmosphere and ocean. Environ Sci Technol, 48(19): 11312-11319, doi:10.1021/es503109p.
Soerensen A L, Skov H, Jacob D J, et al. 2010. Global concentrations of gaseous elemental mercury and reactive gaseous mercury in the marine boundary layer. Environ Sci Technol, 44(19): 7425-7430, doi:10.1021/es903839n.
Soerensen A L, Sunderland E M, Holmes C D, et al. 2010. An improved global model for air-sea exchange of mercury: high concentrations over the North Atlantic. Environ Sci Technol, 44(22): 8574-8580, doi:10.1021/es102032g.
Sørensen A L. 2011. Gaseous mercury in the marine boundary layer: measurements and modeling. National Environmental Research Institute, Aarhus University.
Sprovieri F, Pirrone N, Gärdfeldt K, et al. 2003. Mercury speciation in the marine boundary layer along a 6000 km cruise path around the Mediterranean Sea. Atmos Environ, 37: 63-71, doi:10.1016/S1352-2310(03)00237-1.
Sprovieri F, Pirrone N, Hedgecock I M, et al. 2002. Intensive atmospheric mercury measurements at Terra Nova Bay in Antarctica during November and December 2000. J Geophys Res Atmos, 107(D23): ACH20-1, doi:10.1029/2002JD002057.
Steffen A, Bottenheim J, Cole A, et al. 2013. Atmospheric mercury over sea ice during the OASIS-2009 campaign. Atmos Chem Phys, 13(14): 7007-7021, doi:10.5194/acp-13-7007-2013.
Strode S A, Jaeglé L, Selin N E, et al. 2007. Air-sea exchange in the global mercury cycle. Glob Biogeochem Cycles, 21(1):GB1017, doi: 10.1029/2006GB002766.
Sunderland E M, Mason R P. 2007. Human impacts on open ocean mercury concentrations. Glob Biogeochem Cycles, 21(4): GB4022, doi:10.1029/2006GB002876.
Temme C, Slemr F, Ebinghaus R, et al. 2003. Distribution of mercury over the Atlantic Ocean in 1996 and 1999-2001. Atmos Environ, 37(14): 1889-1897, doi:10.1016/S1352-2310(03)00069-4.
Travnikov, O. 2012. Atmospheric transport of mercury//Liu G L, Cai Y, O’Driscoll N. Environmental Chemistry and Toxicology of Mercury, 331-365, doi:10.1002/9781118146644.ch10.
Travnikov O, Angot H, Artaxo P, et al. 2017. Multi-model study of mercury dispersion in the atmosphere: atmospheric processes and model evaluation. Atmos Chem Phys, 17(8): 5271-5295, doi:10.5194/ acp-17-5271-2017.
Wang F, Saiz-Lopez A, Mahajan A S, et al. 2014. Enhanced production of oxidised mercury over the tropical Pacific Ocean: a key missing oxidation pathway. Atmos Chem Phys, 14(3): 1323-1335, doi:10. 5194/acp-14-1323-2014.
Wang J, Xie Z, Wang F, et al. 2017. Gaseous elemental mercury in the marine boundary layer and air-sea flux in the Southern Ocean in austral summer. Sci Total Environ, 603-604: 510-518, doi:10.1016/j.scitotenv.2017.06.120.
Wang Y Q, Zhang X Y, Draxler R R. 2009. TrajStat: GIS-based software that uses various trajectory statistical analysis methods to identify potential sources from long-term air pollution measurement data. Environ Model Softw, 24(8): 938-939, doi:10.1016/j.envsoft.2009.01.004.
Wängberg I, Schmolke S, Schager P, et al. 2001. Estimates of air-sea exchange of mercury in the Baltic Sea. Atmos Environ, 35(32): 5477-5484, doi:10.1016/S1352-2310(01)00246-1.
Webster J P, Kane T J, Obrist D, et al. 2016. Estimating mercury emissions resulting from wildfire in forests of the Western United States. Sci Total Environ, 568: 578-586, doi:10.1016/j.scitotenv.2016.01.166.
Weiss-Penzias P, Jaffe D, Swartzendruber P, et al. 2007. Quantifying Asian and biomass burning sources of mercury using the Hg/CO ratio in pollution plumes observed at the Mount Bachelor observatory. Atmos Environ, 41(21): 4366-4379, doi:10.1016/j.atmosenv.2007.01.058.
Wikipedia. 2017. Walker circulation. https://en.wikipedia.org/wiki/Walker_circulation [last edited on 2017-08-21].
Xia C, Xie Z, Sun L. 2010. Atmospheric mercury in the marine boundary layer along a cruise path from Shanghai, China to Prydz Bay, Antarctica. Atmos Environ, 44(14): 1815-1821, doi:10.1016/j.atmosenv.2009.12.039.
Yu J, Li B, Sun C, et al. 2015. Total gaseous mercury levels over the Northwestern Pacific Ocean and around Iceland: oceanic, volcanic and geothermal influences. Geochem J, 49(5): 503-512, doi:10.2343/ geochemj.2.0376.
Yue F, Xie Z , Yan J, et al. 2021. Spatial distribution of atmospheric mercury species in the Southern Ocean. J Geophys Res Atmos, 126(17): e2021JD034651, doi:10.1029/2021JD034651.
Yue F, Xie Z, Zhang Y, et al. 2022. Latitudinal distribution of gaseous elemental mercury in tropical western Pacific: the role of the doldrums and the ITCZ. Environ Sci Technol, 56(5): 2968-2976, doi:10.1021/ acs.est.1c07229.

Figure A1 The spatial distributions of SO2column concentrations in the corresponding period of the GEM concentration peaks observed in leg 1–leg 4 in Tropical Pacific.
Figure A2 The linear correlations between the MIT-gcm simulated DGM and observed salinity in leg 1 (a) and leg 2 (b).
21 November 2021;
2 March 2022;
30 March 2022
10.13679/j.advps.2021.0055
: Wang J C, Yue F G, Zhan H C, et al.Features and influencing mechanisms of gaseous elemental mercury over the equatorial Pacific and their differences with the Southern Ocean. Adv Polar Sci, 2022, 33(1): 86-101,doi:10.13679/j.advps.2021.0055
, ORCID: 0000-0003-3629-2247, E-mail: yfg66@ustc.edu.cn
杂志排行

Advances in Polar Science的其它文章
- Foreword
- “Opinion Editorial” category attracts more attention
- One special issue will be published in late 2022
- Potential suitable sites for the calibration of Scientific Echo Sounder in the marginal seas around Antarctica
- Research on submesoscale eddy and front near the South Shetland Islands (Antarctic Peninsula) using seismic oceanography data
- Variability of size-fractionated phytoplankton standing stock in the Amundsen Sea during summer