洋地黄毒苷候选参考方法的建立及评价
2022-07-15唐宁波刘冠财刘晓雨杨丰繁龚志梅孙可其
唐宁波,刘冠财,刘晓雨,杨丰繁,龚志梅,孙可其
(迈克生物股份有限公司参考系统部,四川 成都 611731)
洋地黄毒苷是强心苷类药物,主要用于充血性心力衰竭的治疗。由于洋地黄毒苷作用慢而持久,适用于慢性心功能不全患者长期服用,尤其适用于伴有肾功能损伤的充血性心力衰竭患者[1]。但洋地黄毒苷安全用药范围窄,治疗量接近中毒剂量[2],因此建立洋地黄毒苷的定量检测方法,准确测定人体内洋地黄毒苷的浓度,对临床相关疾病的诊治有重要作用。目前,临床上常用的强心苷类药物检测方法主要有高效液相色谱法、化学发光免疫法、放射免疫法、均相免疫分析法、荧光分析法等[3-4]。在各种方法中,基于免疫学原理的方法具有快速、简便等优点,但易受药物代谢物或与药物结构相似的化合物的影响,导致方法的特异性、准确度和重复性均存在一定的问题[5-6]。因此,为了保证血清洋地黄毒苷检测结果的准确性和可比性,建立可靠的参考方法非常有必要。根据检验医学溯源联合委员会(the Joint Committee for Traceability in Laboratory Medicine,JCTLM)推荐列表,目前国际上有2种测定洋地黄毒苷的参考方法,一种为德国临床化学学会(German Society of Clinical Chemistry,DGKC)建立的同位素稀释液相色谱-质谱法,另一种为德国INSTAND实验室建立的同位素稀释液相色谱-串联质谱法[7]。推荐方法中的前处理步骤较为繁琐,需萃取吹干、复溶、再次吹干、复溶后进样检测,且样本分析时间较长。本研究拟在推荐方法的基础上,建立一种基于同位素稀释超高效液相色谱-串联质谱(isotope dilution ultra high performance liquid chromatography tandem mass spectrometry,IDUPLC-MS/MS)的操作简便、分析时间更短的参考方法,并对所建立的方法进行方法学评价。
1 材料和方法
1.1 仪器和试剂
1.1.1 仪器 UPLC/XEVO TQ-S超高效液相色谱-串联质谱仪(美国Waters公司),ACQUITY UPLC BEH C18色谱柱(1.7 μm,2.1 mm×50 mm,美国Waters公司),十万分之一电子分析天平(德国Sartorius公司),微量移液器(德国Eppendorf公司),Milli-Q超纯水系统(德国Millipore公司),D2012型离心机(北京大龙公司),TD5A-WS型离心机(湖南湘仪公司),YGC-24型氮吹仪(成都雅源科技公司)。
1.1.2 试剂 洋地黄毒苷标准物质(纯度99.0%)购自英国LGC公司,洋地黄毒苷同位素内标(洋地黄毒苷-21,23,23-d3,纯度99.3%)购自加拿大Toronto Research Chemicals公司;叔丁基甲基醚、七水硫酸锌及甲酸铵购自美国Sigma-Aldrich公司,色谱级乙腈和甲酸均购自美国ThermoFisher Scientific公司。
1.2 方法
1.2.1 标准工作液制备 采用重量法配制标准工作液。精密称取洋地黄毒苷标准物质10 mg,用90%乙醇溶液溶解并混匀,配制成20 ng/g的标准工作液,-80 ℃避光保存。
1.2.2 内标工作液制备 内标工作液的制备方法与标准工作液相同。以洋地黄毒苷-21,23,23-d3为内标,用90%乙醇将洋地黄毒苷-21,23,23-d3溶解并混匀,配制成20 ng/g的内标工作液。所有溶液均采用重量法配制,-80 ℃避光保存。
1.2.3 标准曲线制备 取5个5 mL离心管,依次加入50、80、100、120、150 μL标准工作液,然后各加入100 μL内标工作液,使标准品与内标的质量比分别为0.50、0.80、1.00、1.20、1.50。
1.2.4 样本处理 用微量移液器吸取0.1 mL内标溶液至5 mL离心管,精确称得质量,再分别称取适量样本加入内标中混匀,使样本与内标的质量比为1,加入适量水,使最终体积为0.8 mL,震荡混匀,室温下静置平衡1 h,加入0.5 mol/L硫酸锌溶液0.4 mL,沉淀蛋白,充分混匀10 min,2 810×g离心5 min,吸取上清液,加入2.4 mL叔丁基甲基醚溶液萃取,充分混匀10 min,2 810×g离心10 min,吸取上层有机相2.0 mL,45 ℃氮气吹干,加入150 μL流动相复溶,15 100×g离心5 min,吸取上清100 μL,采用ID-UPLC-MS/MS检测。标准溶液与血清样本同时进行处理。
1.2.5 质谱条件 质谱使用电喷雾电离源(electrospray ionization,ESI)正离子模式和多反应监测模式(multiple reaction monitoring,M R M)分析。去溶剂气为氮气,温度为350 ℃,流速为700 L/h;碰撞气为氩气。质谱检测参数见表1。
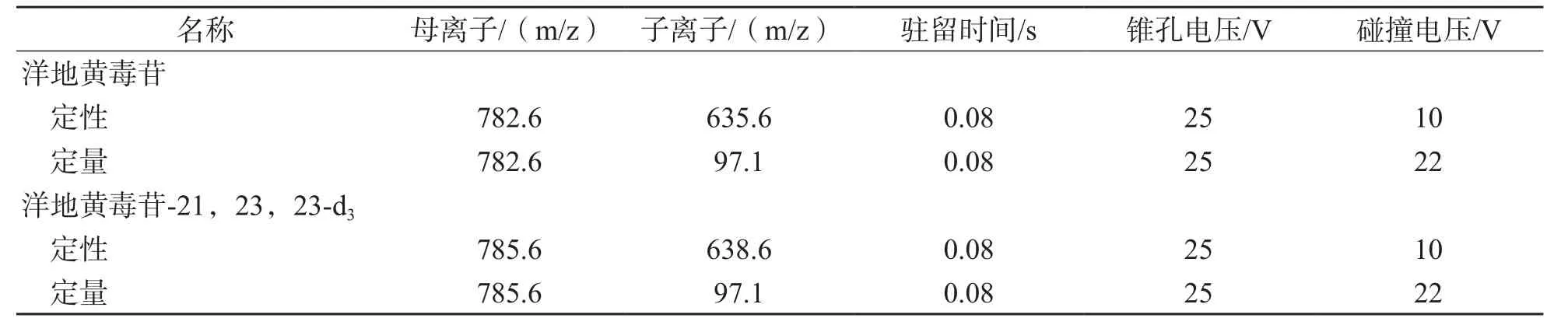
表1 质谱检测参数
1.2.6 色谱条件 色谱柱为ACQUITY UPLC BEH C18色谱柱,进样体积为10 μL,柱温35 ℃,流动相为10 mmol/L甲酸铵(含0.1%甲酸)-乙腈(含0.1%甲酸)(V∶V=40∶60),等度洗脱,流速为0.2 mL/min。
1.2.7 结果计算 所有样本均按照前处理条件作相同处理,每份样本或标准品重复测定3次,计算均值。
1.3 方法学评价
根据美国临床实验室标准化协会(the Clinical and Laboratory Standards Institute,CLSI)C62-A文件[8]、CLSI EP15-A3文件[9]对建立的检测洋地黄毒苷的ID-UPLC-MS/MS方法进行性能评价。
1.3.1 精密度 参考CLSI EP15-A3文件进行批内精密度和批间精密度评价。选择4种不同浓度(A、B、C、D)的冰冻人血清样本,每天分析1批,每批次每个浓度重复测定5次,共测定5 d。根据结果计算出每个浓度的批内变异系数(coefficient of variation,CV)和批间CV。
1.3.2 准确度 准确度验证通过加标回收实验和测定国际能力验证样本进行准确度评价[10]。(1)加标回收。取混合血清,加入适量洋地黄毒苷标准溶液混匀,得到一定质量浓度的洋地黄毒苷混合基础血清,将洋地黄毒苷混合血清分成4份,其中3份分别加入不同量的洋地黄毒苷标准溶液,配制成高、中、低浓度的洋地黄毒苷混合血清样本,将4份血清样本同时进行检测,计算加标回收率。(2)室间比对。测定2020年国际临床化学和检验医学联合会参考实验室外部质量评价计划(the International Federation of Clinical Chemistry and Laboratory Medicine External Quality Assessment Scheme for Reference Laboratories in Laboratory Medicine,RELA)比对样本,每个批次测定2份RELA样本,每份样本重复测定3次,共分析3个批次,计算测量结果与靶值偏移。
1.3.3 检测限与定量限 用0.9% NaCl溶液将已知浓度的高浓度(14.07 nmoL/L)血清样本分别稀释至0.108、0.117、0.141 nmoL/L,每个浓度样本各分成5份,预处理后进行检测,得到实测值。将同时满足偏移≤15%、CV≤20%、信噪比(signal-to-noise ratio,RSN)≥10的浓度值定义为定量下限,将RSN=3时的浓度值定义为检测限。
1.3.4 基质效应 采用基质混合实验的方法计算相对基质效应,即通过检测空白血清基质样本、溶液基质标准溶液、血清与溶液1︰1(V∶V)混合物、血清与溶液8︰2(V∶V)混合物、血清与溶液2︰8(V∶V)混合物这5种基质样本的最终浓度来计算相对基质效应。同时满足偏移≤15%、CV≤15%,则判定为无相对基质效应。
1.3.5 携带污染 评估携带污染时,先重复进样低浓度样本(4.15 nmol/L),然后交替进样高浓度样本(92.00 nmol/L)和低浓度样本。计算2种进样方式下得到的低浓度样本检测结果的差异。若低于预定标准的20%,则认为在测定该高浓度及以下浓度样本时不存在明显的携带污染[11]。
1.3.6 稀释一致性 取洋地黄毒苷浓度超出定量范围的样本(120 nmol/L),稀释到93.30 nmol/L;取洋地黄毒苷定量范围内的样本(10 nmol/L)分别稀释到3.97 nmol/L和1.98 nmol/L。分别检测定量范围外的稀释样本和定量范围内的稀释样本,结果应同时满足回收率100%±15%,CV<15%。
1.3.7 特异性 用建立的ID-UPLC-MS/MS方法同时测定洋地黄毒苷及与其结构相似的化合物地高辛,若地高辛与洋地黄毒苷达到色谱分离,且响应值无明显变化,则说明洋地黄毒苷检测特异性好,不受结构相似化合物的影响。
1.3.8 稳定性 评估洋地黄毒苷在生物基质中的稳定性。取2份血清样本,分别均分为4等份,一份在常温放置4、8、12、24 h,另一份在2~8 ℃下放置1、2、3、4 d,然后进样检测,观察样本在常温及2~8 ℃下的稳定性;为了评估样本处理后的稳定性,将处理后的样本在室温和进样器中分别放置4、8、12、24 h后进行检测。若测定结果的均值在标示值±15%范围内,则判定为满足稳定性要求[12]。
1.4 不确定度评定
依据CNAS-GL006:2019文件[13]对血清样本检测结果不确定度的来源进行分析。参照《测量不确定度评定与表示指南》[14](Guide to the Expression of Uncertainty in Measurement,GUM)和《分析测量中不确定度的量化》[15](Quantifying Uncertainty in Analytical Measurement,QUAM)对结果进行不确定度评定。其中,A类不确定度主要来自样本的重复测定;B类不确定度主要来自洋地黄毒苷标准物质纯度、洋地黄毒苷标准物质称量、移液器移液过程引入的不确定度。
B类不确定度合成公式为:
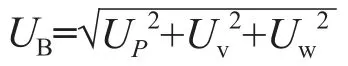
式中UP、Uv和Uw分别为标准物质纯度、样本称量以及移液器引入的不确定度。合成不确定度公式为:
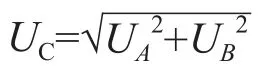
扩展不确定度计算公式为:
Ue=k×UC
式中k为包含因子(k=2)。
2 结果
2.1 标准曲线及色谱图
以标准品与内标的质量比和对应峰面积比值制作标准曲线,方程为Y=1.162 8X+0.014 4(r2=0.999 7),见图1。洋地黄毒苷标准溶液和内标的色谱图见图2。洋地黄毒苷的线性范围为2.8~94.9 nmol/L。
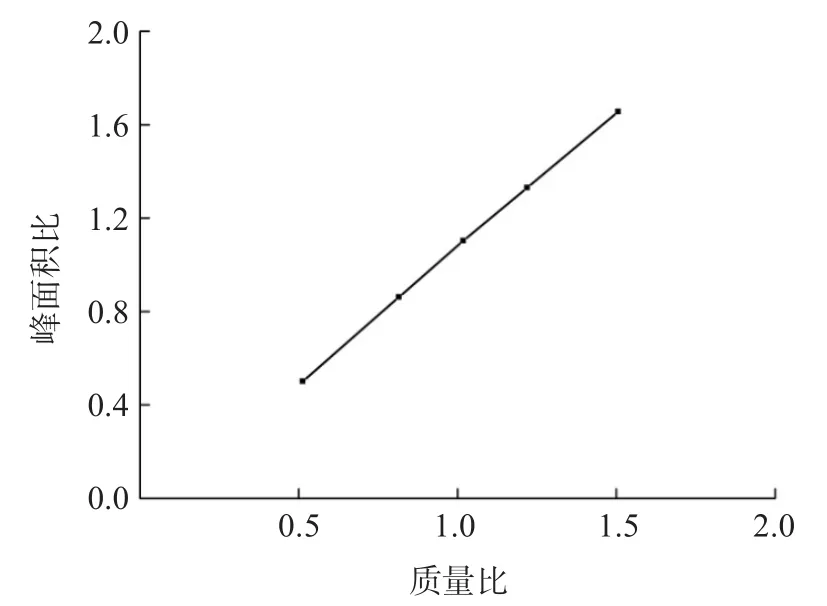
图1 洋地黄毒苷标准曲线

图2 洋地黄毒苷和内标色谱图
2.2 方法学评价
2.2.1 精密度 ID-UPLC-MS/MS测定A、B、C、D 4个水平的洋地黄毒苷批内精密度≤1.98%,批间精密度≤1.06%。见表2。
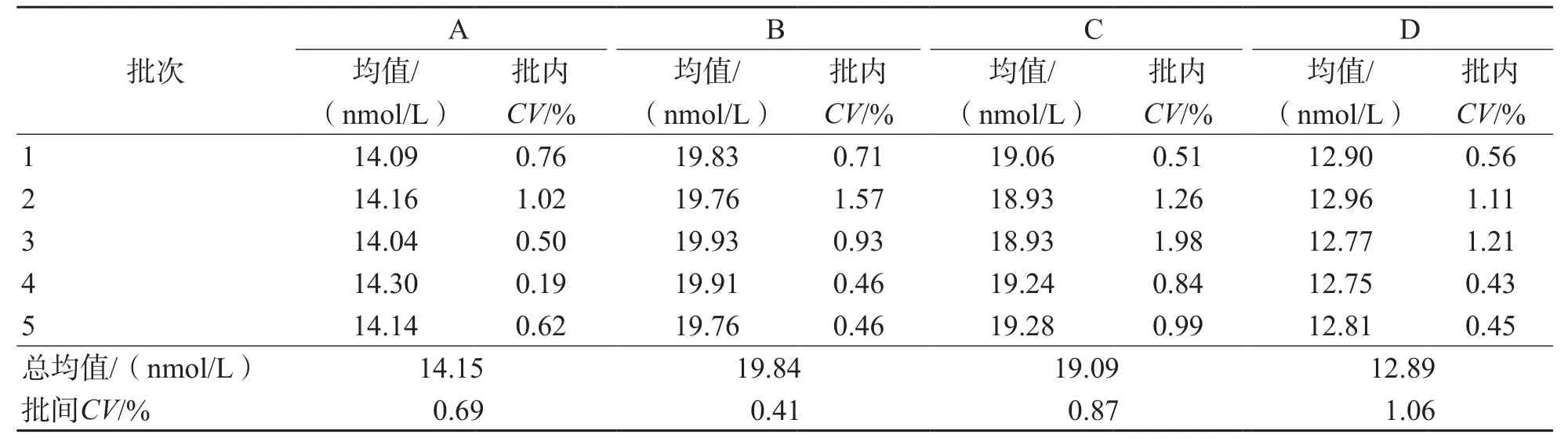
表2 ID-UPLC-MS/MS检测血清洋地黄毒苷的精密度
2.2.2 准确度 ID-UPLC-MS/MS测定血清洋地黄毒苷的加标回收率为100.03%~100.48%。重复测定3次,CV≤1.80%,见表3。参加2020 RELA室间比对,本实验室2020 RELA-A、2020 RELA-B样本检测结果与总均值的偏移分别为0.62%、0.29%。

表3 加标回收率测定结果
2.2.3 检测限与定量限 采用ID-UPLC-MS/MS测定血清洋地黄毒苷,同时满足偏移≤15%、CV≤20%、RSN≥10的浓度为0.095 nmol/L,将其定义为定量下限;检测限为0.032 nmol/L。见表4。
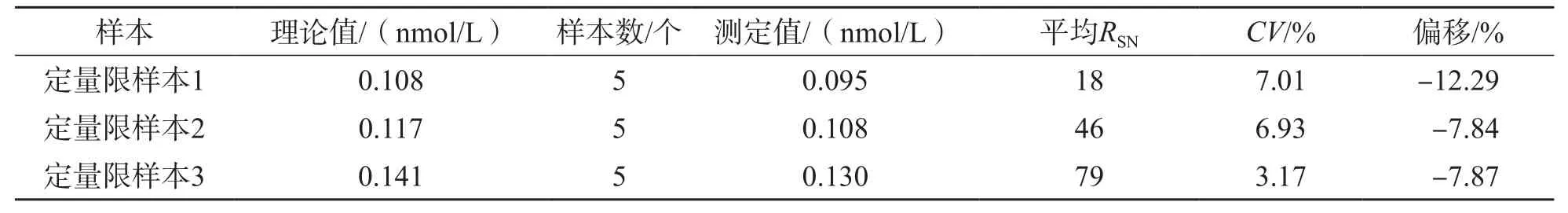
表4 洋地黄毒苷的定量限评估结果
2.2.4 基质效应 5种基质样本测定值与理论值同时满足偏移≤15%、CV≤15%,判定为无相对基质效应。见表5。
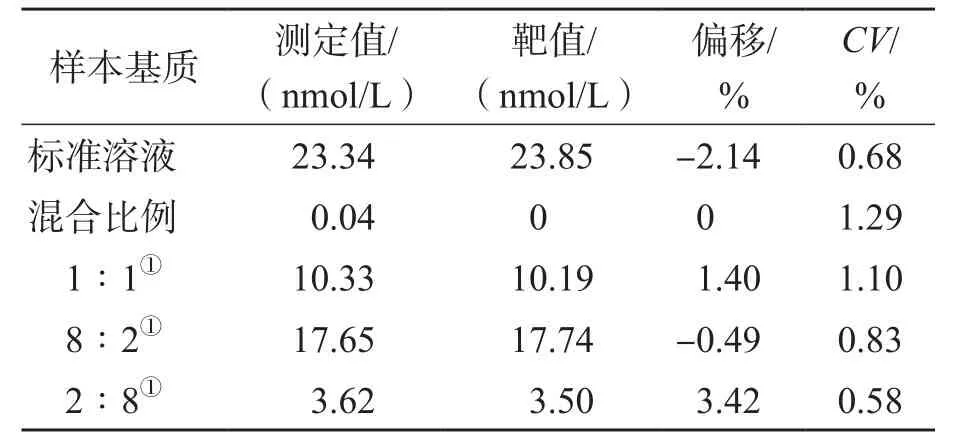
表5 洋地黄毒苷的基质效应评估结果
2.2.5 携带污染 ID-UPLC-MS/MS检测洋地黄毒苷低浓度样本重复进样时的均值为4.15 nmol/L,交替进样测得低浓度样本的均值为4.19 nmol/L,计算得到携带污染率为0.88%。
2.2.6 稀释一致性 2份定量范围内的样本(样本1、样本2)和1份定量范围外的样本(样本3)测定结果与靶值偏移均<2%。见表6。该方法满足稀释一致性要求。
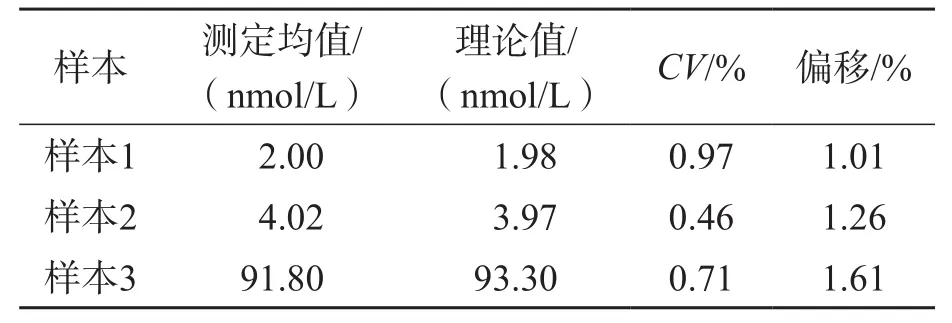
表6 洋地黄毒苷的稀释一致性估结果
2.2.7 特异性 洋地黄毒苷和地高辛可实现基线分离,并且响应值与加入地高辛前比较,无明显变化。见图3。
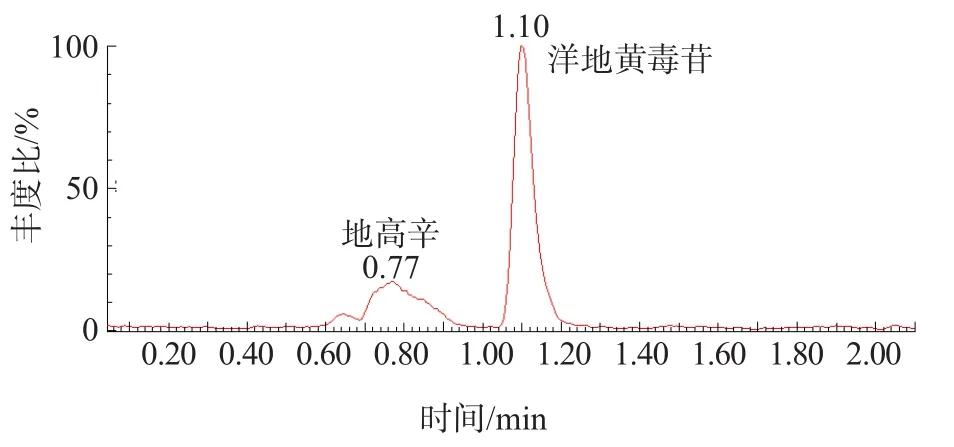
图3 洋地黄毒苷及地高辛检测色谱图
2.2.8 稳定性 血清样本在室温下放置4、8、12、24 h及2~8 ℃下放置1、2、3、4 d的测定结果偏移在±1.91%以内,见表7。血清样本处理后在室温下和自动进样器中放置4、8、12、24 h的测定结果偏移在±1.56%以内,见表8。所有测定结果均在标示值±15%以内,满足稳定性要求。
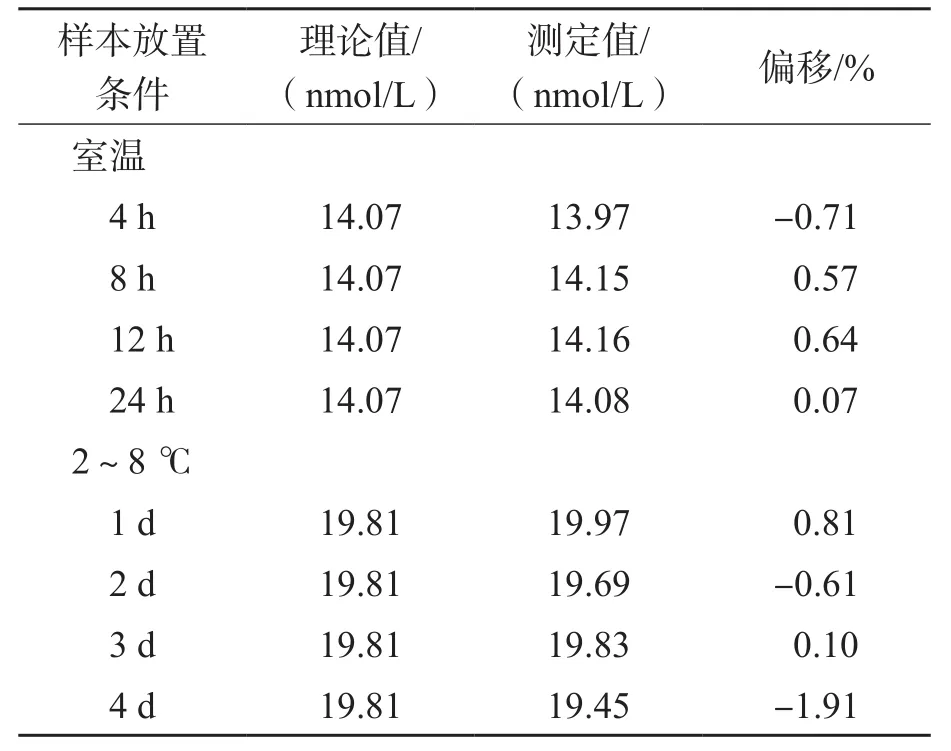
表7 未作前处理的血清样本在不同样本保存条件下的稳定性
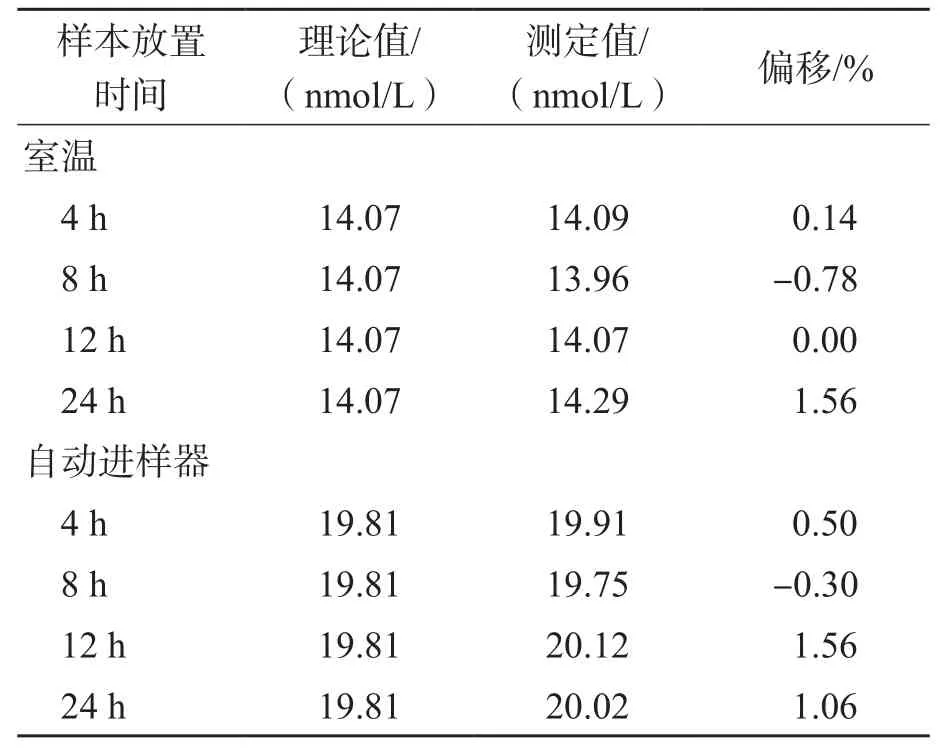
表8 处理后的样本的稳定性
2.3 不确定度评定
以2020 RELA-A和2020 RELA-B样本为例,对ID-LC-MS/MS检测血清洋地黄毒苷的不确定度进行评定,不确定度评定结果见表9。根据表9中的相对不确定度分量计算2020 RELA-A和2020 RELA-B样本的相对扩展不确定度(包含因子k=2)分别为2.47%、2.11%。2020 RELA-A和RELA-B样本测定值分别为14.16、19.87 nmol/L,扩展不确定度分别为±0.35 nmol/L、±0.42 nmol/L(包含因子k=2)。
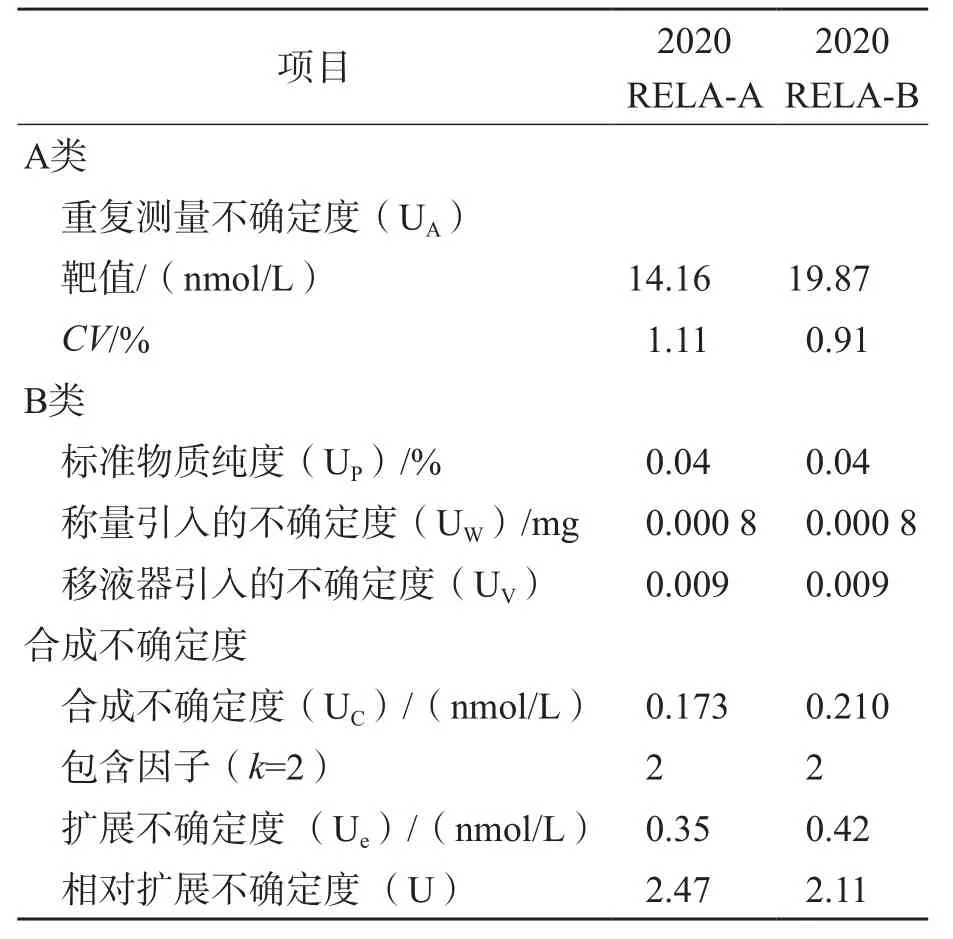
表9 不确定度评定结果
3 讨论
JCTLM推荐了2种液相色谱-串联质谱检测洋地黄毒苷的参考方法,其最大的亮点是使用Cs+作为加合离子。当使用Cs+作为加合离子时,可以提高该方法的特异性,不会受到生物来源样本中其他物质的干扰,但使用的化合物氯化铯有毒,不建议使用,且我国的研究中几乎没有关于Cs+的使用方法及浓度范围。德国INSTAND实验室建立的洋地黄毒苷参考方法,样本前处理需先调节样本的pH值,再使用叔丁基甲基醚-乙酸乙酯(V∶V=1∶1)进行萃取,氮气吹干,采用甲醇复溶,再次进行氮气吹干,使用流动相复溶进样,前处理过程中的2次复溶吹干,有利于除去样本中洋地黄毒苷的干扰物质,对峰型图有一定的改善作用,但是会增加样本的前处理时间,使得样本前处理变得繁琐,且样本分析时间为15 min,相对较长;同时,国内外关于液相色谱-串联质谱法用于洋地黄毒苷检测的研究很少。因此,本实验室在基于以上因素和保证方法准确度以及精密度的前提下,尝试建立一种ID-UPLC-MS/MS测定血清洋地黄毒苷的候选参考方法,通过对色谱、质谱方法及样本前处理进行摸索与优化,经蛋白沉淀、液液萃取、吹干、复溶即可进样分析,大大缩短了样本前处理时间,且样本分析时间缩短至4 min,有效提高了该方法的实用性和可操作性,可用于常规检测系统的量值溯源及准确度评价。
本研究在进行质谱条件摸索时,发现[M+NH4]+在甲酸铵溶液体系中会形成信号更强的分子离子峰[16]。与JCTLM推荐方法中的母离子[M+Cs]+比较,[M+NH4]+中的NH4+为实验室常用离子,其溶液浓度基本固定,对仪器管路影响较小,而Cs+很少有实验室会用到,溶液配制浓度不确定,未知溶液浓度对仪器以及实验结果有较大影响,故本研究流动相选择甲酸铵溶液。在色谱条件中,本研究未采用推荐方法的梯度洗脱,而是选择等度洗脱,在保证色谱峰型较好的前提下,减少了样本分析时间。
洋地黄毒苷是属于强心苷类药物,与之相似的药物还有地高辛,洋地黄毒苷与地高辛空间结构相似,地高辛结构的C14号位置比洋地黄毒苷多1个羟基,其他结构均一致,在检测洋地黄毒苷时,有可能会受到地高辛的干扰。本研究干扰实验结果显示,洋地黄毒苷和地高辛可实现基线分离,并且色谱峰响应值在加入地高辛前后无明显变化,说明建立的ID-UPLC-MS/MS方法检测洋地黄毒苷不受结构相似物质的干扰。
本研究方法学评价结果显示,ID-UPLC-MS/M S检测洋地黄毒苷的线性范围为2.8~94.9 nmol/L,不同基质样本对检测结果无明显基质效应影响,定量限为0.095 nmol/L,检测限为0.032 nmol/L,携带污染率为0.88%,批内精密度≤1.98%,批间精密度≤1.06%,加标回收率为100.03%~100.48%。重复测定的CV≤1.80%,2020 RELA样本检测结果与总均值的偏移≤0.78%。
综上所述,本研究建立的ID-UPLC-MS/MS候选参考方法的准确度高、精密度好、特异性强、操作简便,不受其他生物来源的物质干扰,且操作简便,分析时间较短,可用于洋地黄毒苷常规方法的准确度评价。
