Phylogenetic Analysis of Morels Based on ITS and LSU Sequences
2022-07-13SenlinZHUBoCHENFenJIANZhunXIANGQianLUOXiaominWANGXuCHENBangxiZHANG
Senlin ZHU Bo CHEN Fen JIAN Zhun XIANG Qian LUO Xiaomin WANG Xu CHEN Bangxi ZHANG

Abstract[Objectives] Fifty two strains were identified and phylogenetically analyzed to solve the current phenomenon of "synonym and homonym" in morels. [Methods]Based on the ITS and LSU sequences of ribosomal DNA, the collected 52 morel samples were sequenced, and BLAST alignment was performed by NCBI to construct phylogenetic trees. [Results] The collected morel strains were mainly divided into two major groups: namely black morel and yellow morel groups, of which the black morel group was most, mainly including Morchella sextelata, Morchella importuna, Morchella crassipes, and Morchella eximia. There were very few yellow morel strains, including M. crassipes and M. eximia. [Conclusions]This study lays a foundation for the classification and research of morel germplasm resources, and provides a powerful reference value for the research and production of highquality morel strains.
Morchella Dill.ex Pers.:Fr., belongs to Morchellaceae in Pezizales of Pezizomycetes in Ascomycota[1]. Because it is quite similar to sheeps belly with honeycombshaped surface which often domes or spires, it is named morel mushroom. In different parts of China, people have given it different names, such as sheep stomach and sheep stomach dish[2]. Morels taste crisp, tender and delicious with unique flavor, contains high vitamin and essential amino acids, and have great edible value[3]. Studies have shown that morels not only contain a variety of antibacterial and antiviral active ingredients, but also are rich in polysaccharides that can inhibit tumors and have many effects such as regulating immune function and resisting fatigue[4]. Morels have significant effects in nourishing the kidney, strengthening yang, nourishing the brain, and refreshing, and are called a natural "hormone"[5]. Therefore, Morel mushroom has become a worldrecognized precious and rare edible and medicinal fungus[6]. It has shown great development potential in the fields of food, medicine, health care, and cosmetics[7].
In recent years, with the recognition of morels efficacy, more and more people have invested in morels commercial cultivation, which has become a pillar industry which consolidates the achievements of poverty alleviation and effectively links rural revitalization. However, due to the great plasticity and artificiality of its morphological characteristics, problems such as confusion of strains and uncertain classification of germplasm resources in the market are more prominent, such as lack of research on germplasm resources, serious species degradation, synonym and homonym[8], which increases the risk of morel cultivation[9], and brings certain difficulties to the healthy development of the morel industry. With the development of molecular biology and the development of the morel multigene sequence model database MLST[10] (multilocussequence typing), many molecular marker techniques are widely used in the molecular identification of morel. ITS (ribose rDNA internal transcribed spacer) sequence and LSU (ribosomal large subunit) sequence analysis have the advantages of excellent specificity, high accuracy and simple operation, and have been widely used. In this study, 52 morel samples were collected and sequenced for the ITS and LSU sequences, which were subjected to BLAST alignment through NCBI and used for the construction of phylogenetic trees, aiming to solve the current phenomenon of "synonym and homonym" in morels. This study lays a theoretical foundation for the healthy development of the morel industry.
Materials and Methods
Materials
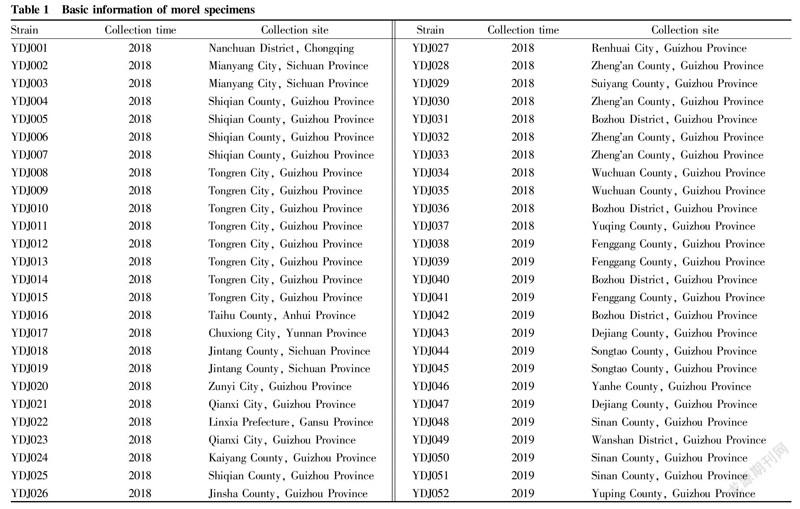
The collected 44 cultivars and 8 wild morel strains (strain numbers YDJ008YDJ0015) were used as test materials. The strain numbers and sources are shown in Table 1.
Methods
DNA extractionDNA was extracted using the Novel Plant Genomic DNA Kit following the instructions in the kit.
PCRThe primers ITS1: 5TCCGAGGTGAACCTGCGG3, ITS4: 5TCCTCCGCTTATTGATAT3 were used as specific fragments to amplify the ITS region. With primers LSU1: 5′TATCACTAAGCGGAGGAAAAGA3′and LSU4: 5′AGGGAACCAGCTACTAGATGG3′ as primers, PCR was performed according to the Gao Shans method[11].
Sequence determinationPCR products were directly sequenced. After the samples were purified, Sangon Biotech (Shanghai) Co., Ltd. was entrusted to conduct bidirectional sequencing, and the complete sequences were obtained by splicing with Contig Express 10.0 software.
Construction of phylogenetic treeThe morel nucleotide sequences obtained by sequencing were subjected to Blast alignment with the NCBI database, and phylogenetic trees was constructed by the neighborjoining method (NJ). The validation parameter was Bootstrap=1 000.
Results and Analysis
Acquisition of ITS and LSU gene fragments
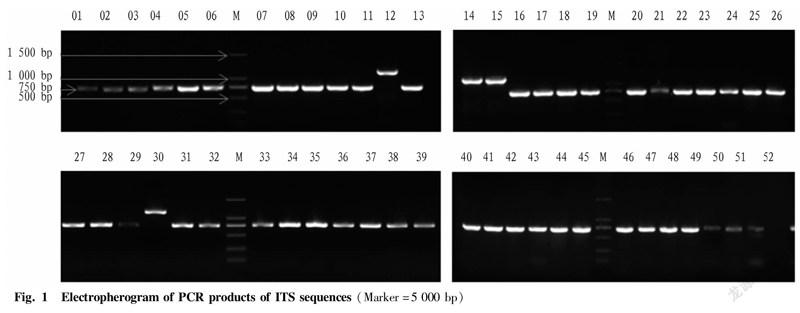
ITS and LSU sequences were amplified using primers ITS1, ITS4, LSU1 and LSU4. The collected total morel DNA was amplified by PCR and detected by agarose gel electrophoresis (Fig. 1). The results showed that the ITS sequences of the samples numbered YDJ012 and YDJ030 are the longest, about 1 200 bp, while the ITS sequences of the samples of YDJ014 and YDJ015 are second only to the samples of YDJ012 and YDJ030, about 1 000 bp, and the bands of other samples are around 750 bp. The sequences amplified by LSU primers are shown in Fig. 2 below. All sample bands have the same size and are distributed between 500-750 bp, which is consistent with the results reported by WIPF.
Phylogenetic analysis based on ITS sequences
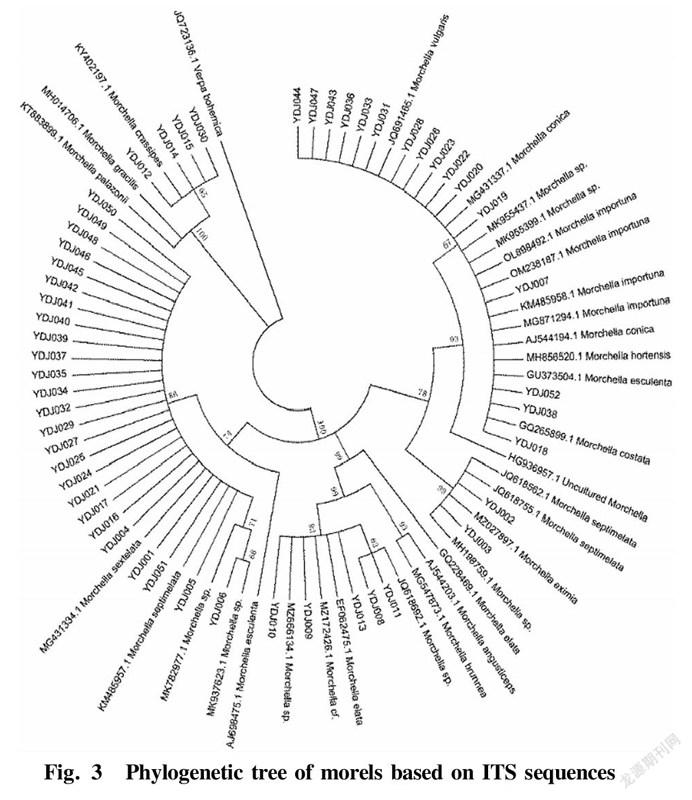
The 52 sequences of the tested materials, the sequences with high homology downloaded from the database and the sequences of other species of Morchella that were not involved in the experimental materials were used to construct a phylogenetic tree by the neighborjoining method with Verpa bohemica as the outgroup, and the displayed evolutionary tree is shown in Fig. 3.
It can be seen from the phylogenetic tree that with Verpa bohemica as the outgroup, the 52 samples could be divided into 5 major groups. Sixteen morel samples including YDJ007, YDJ018 and YDJ019 and Morchella importuna (KX809735.1) were clustered in group I with the Bootstrap value of 99%, indicating that the 16 morel strains were closely related. Two morel samples numbered YDJ002 and YDJ003 were relatively small in sequence and relatively closely related. They were clustered in group II together with Morchella eximia (KM587980.1) with a Bootstrap value of 99%. Five morel samples including YDJ008, YDJ009 and YDJ010 were clustered into group III with Morchella elata (EF08996.1) with a Bootstrap value of 93%. Twenty five morel samples including YDJ001, YDJ004 and YDJ005 were relatively small in sequence and relatively closely related, and clustered with different species represented by Morchella sextelata and Morchella septimelata from the database into group IV with a Bootstrap value of 86%. The above four groups all belonged to the black morel class group, while the four samples numbered YDJ012, YDJ014, YDJ015 and YDJ030 were clustered with Morchella crassipes (KY402197.1) with a Bootstrap value of 100% into group V, belonging to the yellow morel class group. In summary, based on the phylogenetic tree of the 52 morel samples based on ITS sequences, they could be classified into two taxonomic groups, namely the black morel group and the yellow morel group.
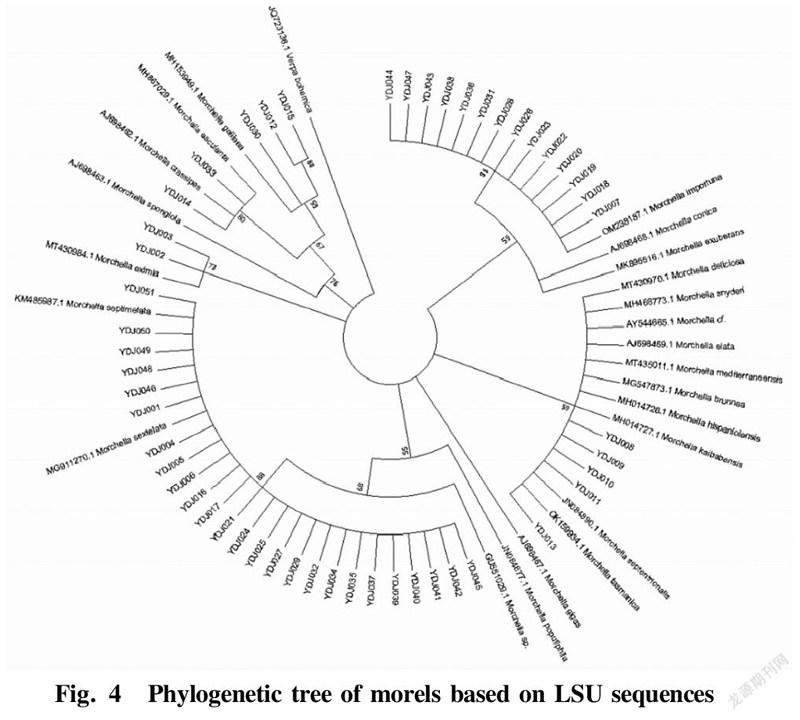
Phylogenetic analysis based on LSU sequences
The LSU sequences of the 52 test materials were subjected to Blast alignment in the NCBI database, and the sequences with higher similarity were downloaded. A phylogenetic tree was constructed using MEGA7.0 software by the NJ algorithm, as shown in Fig. 4. From the phylogenetic tree, it can be seen that the 52 samples could be divided into 4 major groups. The LSU sequences of 16 morel samples including YDJ007, YDJ018 and YDJ019 were clustered into group I with M. importuna (JX171380.1) and M. importuna (MG871328.1) with a Bootstrap value of 99%, indicating that the LSU sequences of these 16 morel strains were very similar to that of M. elata. The five morel samples of YDJ008, YDJ009, YDJ010, YDJ011 and YDJ013 were clustered into group II with M. elata (AJ698469.1) with a Bootstrap value of 59%, indicating that the LSU sequences of these five morel samples were very similar to that of M. elata. Twenty five morel samples including YDJ001, YDJ004 and YDJ005 were clustered together with M. sextelata (MG911270.1) and M. septimetata (KM485987.1) into group III with a Bootstrap value of 88%, indicating that the LSU sequences of these 25 morel strains were very similar to M. sextelata and M. septimetata. Two morel samples numbered YDJ002 and YDJ003 were clustered on a branch with the Morchella eximia (MT430984.1) with a Bootstrap value of 73%.
The five wild morel samples of YDJ008, YDJ009, YDJ010, YDJ011 and YDJ013 were clustered on the branch of M. elata, indicating that the LSU sequences of these five morel samples were very similar to that of M. elata, and they all belonged to the black morel group. The 4 samples numbered YDJ012, YDJ014, YDJ015 and YDJ030 were clustered with M. crassipes (AJ698462.1) into group IV with a Bootstrap value of 67%, while 12, 15 and 30 were clustered with M. spongiola into a group with a Bootstrap value of 76%, indicating that the LSU sequences of these four morel samples were very similar to M. crassipes and M. spongiola, and they belonged to the yellow morel group. To sum up, through the phylogenetic analysis of the LSU sequences of the 52 morel samples, they could be classified into two taxonomic groups: black morels and yellow morels.
Discussion
ITS and LSU sequences are often used to identify the relatives of fungi. The ITS sequence is the region located in the gaps of 18S, 5.8S and 28S in rDNA, called the internal transcribed spacer, which is part of the nontranscribed region of the fungal rRNA gene. It has the advantages of fast evolution and moderate fragment length[12]. The ITS sequence is often used for taxonomic identification and phylogenetic studies at the genus level and the levels below genus[13]. The large ribosomal subunit of LSU is the large ribosomal subunit sequence, which is an RNA sequence located at 28S, and the sequence is relatively conservative. It is widely used in the study of the relationship between the components at the levels below genus and between the members of higher taxonomy[11]. In this study, morel germplasm resources were collected for ITS and LSU sequence determination and analysis, and relatively complete phylogenetic trees were constructed by the NJ algorithm. Regardless of the results of the phylogenetic tree constructed by the amplified fragments of ITS or the amplified fragments of LSU, the collected morel samples could be assigned to corresponding groups. However, using LSU sequences for phylogenetic analysis, the division of the collected samples numbered YDJ012, YDJ014, YDJ015, YDJ030 and YDJ033 could be finer and more convincing. Therefore, the selection of ITS sequences and LSU sequences for phylogenetic analysis can effectively solve the confusion of morel strains due to synonym and homonym. This study lays a foundation for the classification and research of morel germplasm resources, and provides a strong reference value for the research and production of highquality morel strains.
References
[1] HIBBETT DS, BINDER M, BISCHOFF JF, et al. A higherlevel phylogenetic classification of the fungi[J]. Mycological Research, 2007(111): 509-547.
[2] DU XH, ZHAO Q, YANG ZL. Diversity, evolutionary history and cultivation of morels: A review[J]. Mycosystema, 2014, 33(2): 183.
[3] DAI YC, YANG ZL. A revised checklist of medicinal fungi in China[J]. Mycosystema, 2008(6): 801-824. (in Chinese).
[4] CHEN XD, ZHU R, LAN J. Advances in the study of Morchella[J]. Acta Edulis Fungi, 2002(2): 56-61. (in Chinese).
[5] XU YQ, ZHANG MS, ZHANG LX. Biological characteristics, nutritional value and cultivation techniques of Morchella[J]. Seed, 2006(7): 97-99.
[6] MAO XL. Chinese mushroom[M]. Beijing: Science Press, 2009. (in Chinese).
[7] ZHANG GL, ZHANG WM, LI G. Research and utilization of Morchella[J]. Chinese Wild Plant Resources, 1999(1): 1-4. (in Chinese).
[8] DU XH, ZHAO Q, YANG ZL. Research progress on diversity, evolution history and cultivation of Morchella[J]. Mycosystema, 2014, 33(2): 183-197. (in Chinese).
[9] ZHAO XX. Morels in northern Guizhou[J]. Edible Fungi, 1988(4): 9. (in Chinese).
[10] DU XH, ZHAO Q, ODONNELL K, et al. Multigene molecular phylogenetics reveals true morels (Morchella) are especially speciesrich in China[J]. Fungal Genetics & Biology Fg & B, 2012, 49(6): 455-469.
[11] GAO S. Studies on phylogenetic relationships of Pleurotus based on nuclear large subunit ribosomal DNA and its sequences[D]. Wuhan: Huazhong Agricultural University, 2008: 15-17. (in Chinese).
[12] BRANS TD, SHEFFERSON RP. Evolutionary studies of ectomycorrhizal fungi: Recent advances and future directions[J]. Can J Bot, 2004, 82(8): 1122-1132.
[13] MA YS, FEI DL, WANG WJ, et al. ITS sequence analysis of several powdery mildew from Henan Province[J]. Journal of Henan Agricultural Sciences, 2012, 41(9): 87-90. (in Chinese).
Editor: Yingzhi GUANG Proofreader: Xinxiu ZHU
杂志排行

农业生物技术(英文版)的其它文章
- Report on the Breeding of Dahen 799 Broilers
- Evaluation and Analysis for Survey of Tea Production Quality Safety Management and Control
- Study on the Preparation of "Oil-tea" Instant Tea from the Compound Extract of Green Tea and Ginger
- Research Progress on Chemical Constituents and Pharmacological Effects of Zhuang Medicine Cocculus laurifolius DC.
- Study on Quality Standard of Lujing Yiqi Shengxue Pills
- Research on the Development of Guangxi Zhuang and Yao Ethnic Medicine Industry