Anchoring metal ions in amine-functionalized boron imidazolate framework for photocatalytic reduction of CO2
2022-07-11GuilnXuQinLongHongYyongSunMengLiuHiXiZhngJinZhng
Guiln Xu,Qin-Long Hong,Yyong Sun,Meng Liu,Hi-Xi Zhng,∗,Jin Zhng,∗
a State Key Laboratory of Structural Chemistry,Fujian Institute of Research on the Structure of Matter,Chinese Academy of Sciences,Fuzhou 350002,China
b Key Lab for Sport Shoes Upper Materials of Fujian Province,Fujian Huafeng New Material Co.,Ltd.,Putian 351164,China
Keywords:Metal-organic framework CO2 reduction Boron-imidazolate framework Photocatalysis Structure
ABSTRACT The photocatalytic reduction of CO2 to energy-rich chemicals is highly appealing for alleviation of energy crisis and environment pollution.The introduction of different active sites is a key factor to determine the reaction activity and selectivity.Here,we demonstrate the metal ion-dependent performance for photocatalytic CO2 reduction by anchoring transition metal ions (Co2+ and Ni2+) in an amine-functionalized boron imidazolate framework (BIF-43).As a result,Ni@BIF-43 realized a high selectivity of 90.2% for the CO2-to-CO,while Co@BIF-43 achieved more efficient conversion with a high CO production rate of 2036.0 μmol g−1 h−1.Significantly,precise control of isolated metal site on a well-defined structure through coordination-assisted strategies enables us to better understand the specific effects of different metal-ion species on photoreduction of CO2 as well as the catalytic mechanism.
Excessive emission of carbon dioxide (CO2) from the burning fossil fuels results in serious global climatic change and energy shortage [1–4].Currently,the photocatalytic conversion of CO2to valuable energy-rich chemicals has been considered as one of promising technologies to alleviate above issues [5–8].Unfortunately,owing to the high thermodynamic stability of CO2molecule,the activation and reduction of CO2are very difficult and complex[9,10].The multiple proton coupled electron transfer process is required to CO2photoreduction,which is accompanied by the competing hydrogen evolution reduction (HER) leading to a wide variety of products [11].In the past several decades,a series of efficient photocatalysts based on semiconductors (such as TiO2,CdS,g-C3N4) and nanocomposites have been developed [12–22].Due to the complicated structural components of catalysts and the large number of possible products,fundamental understanding the dependence between the structure and composition of catalyst on the activity and selectivity of CO2photoreduction is a significant challenge.
Loading active metal atoms/ions onto heterogeneous supports is a very versatile route to maximize the fraction of active sites and tailor properties of catalysts [23,24].The transition-metal (TM) catalyst has attracted worldwide attention in catalytic field.Especially,the metal Ni and Co serving as active sites play important roles in catalytic reaction [5,7,13,19].Generally,active carbon,metal oxides,silica and zeolites are the most common supports,because of their effective voids and defect sites [25–31].However,it is difficult to precisely control the location and the local environment of metal atoms/ions on these mostly disordered anchoring surfaces,easily resulting in nonuniform distribution as well as complicated metal–support interactions.Compared with the conventional inorganic supports,metal-organic frameworks (MOFs) possess many attractive features such as well-defined structure,high surface area,tunable inorganic nodes and organic linkers,and rich surface coordination sites,and have thus emerged as possible supporters to embed catalytic metal sites [32–36].Introducing TM atoms into a MOF matrix could open up great opportunities to tune the electronic properties of TMs as CO2RR active sites and maintain relatively simple atomic coordination for fundamental mechanism study.Recently,many studies have shown that MOFs can serve as functional supports for anchoring external metal sites on secondary building units (SBU) or organic struts [37–41].However,the reaction condition for photocatalytic reduction of CO2is somewhat harsh,and it is urgently needed to develop new porous and stable functional MOFs.
Boron imidazolate frameworks (BIFs),a kind of MOFs constructed by the crosslinking of pre-synthesized boron imidazolate ligands and metal ions,are unexpectedly found to show improved chemical stability due to the presence of robust M-N coordination bonds and the strong covalent B-N bonds [42].Recently,some BIFs have been used as catalysts to reduction CO2study,which offered accurate insight into the catalytic processesviaadjusting the local environment of metal nodes [43–47].However,it is difficult to anchor metal on the organic struts in BIFs because of the lack of uncoordinated functional groups,such as −COOH,−OH,−NH2.Consequently,the construction of BIF-based catalyst with a special binding microenvironment to provide a new direction to functionalize BIFs is quite desirable.
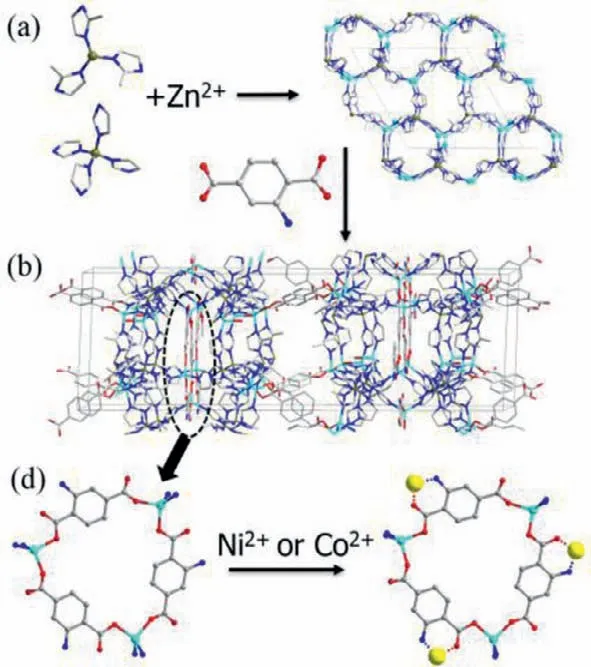
Fig.1.Structural analysis of BIF-43.(a) The 2D boron imidazolate layer linked by BH(mim)3−,B(im)4−and metal ions.(b) 3D structure of BIF-43.(c) Details of the Zn3(2-ATP)3 ring.
Here,we designed an amine-functionalized boron imidazolate framework (BIF-43) which was used as a support to stabilize the transition metal ions (Ni2+and Co2+) in pores,obtaining Ni@BIF-43 and Co@BIF-43.By anchoring external metal ions,it offered a simple and precise structural model to analyze the metal nodesdependent effect on photoreduction of CO2.As expected,the BIF-43 with different catalytic active centers exhibit diverse effects on the activity and selectivity of mainly reductive products.It is worth noting that Ni@BIF-43 showed very high catalytic selectivity(90.2%) for CO,which has far exceeded the selectivity of Co@BIF-43 (64.1%).The close contact between adsorption sites and active sites effectively activates CO2molecules and improves photocatalytic conversion of CO2.This strategy offered us an important insight to prepare BIF-based materials for CO2capture and photoreduction.
BIF-43 (Zn11[B(im)4]6[BH(mim)3]2(2-ATP)6(OH)2,im=imidazole,mim=2-methylimidazole,2-ATP=2-aminoterephthalic acid) was successfully designed and synthesized through the selfassembly of Zn(CH3COO)2·2H2O and three different ligands KBH(2-mim)3,KB(im)4and 2-ATP (summary of crystal data and structural refinements are shown in Table S1 in Supporting information).It is the first example that integrates both tetradentate B(im)4−ligand and tridentate BH(mim)3−ligand.Despite such a complex composition,BIF-43 can be simply considered to be built from the 2D boron-imidazolate-zinc layer pillared by the 2-ATP ligands.The single-crystal X-ray diffraction analysis exhibits that BIF-43 crystallizes in the hexagonal space groupP62c and features a 3D pore structure.There are three crystallographically independent tetrahedral zinc sites in the asymmetric unit.Each Zn1 is bonded to three N from two B(im)4−and one BH(mim)3−,and one O atoms from 2-ATP ligand (Fig.S1 in Supporting information).Each Zn2 is bonded to three N from three B(im)4−and one charge-balancing OH−(Fig.S2 in Supporting information).Zn1 and Zn2 ions are bridged by B(im)4−and BH(mim)3−ligands to form ahcblayer parallel to theabplane (Fig.1a).Two such layers are bridged by Zn3 ions to form a 2D sandwich-like layer,with Zn3(2-ATP)3rings at the center of the sandwich layer (Figs.1b and c and Fig.S3 in Supporting information).Furthermore,these sandwich layers are linked by 2-ATP ligands that bonded on Zn1 to yield a 3D porous structure (Fig.1b).Its phase purity,thermal and chemical stability were confirmed by powder X-ray diffraction (PXRD) and thermogravimetry analysis (TGA) (Figs.S4-S7 in Supporting information).As estimated by the PLATON program,the free space in BIF-43 is 5459.2 ˚A3per unit cell with the pore volume ration of 43.0%.N2sorption measurements (Fig.S8 in Supporting information) at 77 K exhibit type-I isotherms behavior with the calculated Brunauer-Emmett-Teller(BET) and Langmuir surface areas of 539.8 and 779.3 m2/g,respectively.Moreover,BIF-43 shows a high affinity for CO2due to the presence of −NH2in the pore [48,49].The CO2uptake values are 97.2 cm3/g at 273 K and 63.9 cm3/g at 298 K under 1 atm (Fig.S9 in Supporting information).The adsorption enthalpy (Qst) near the zero coverage was calculated to be 32.4 kJ/mol,which was significantly higher than those reported BIFs without −NH2groups [50].The stronger affinity of BIF-43 toward CO2is more conductive to the activation of CO2during the reduction process.
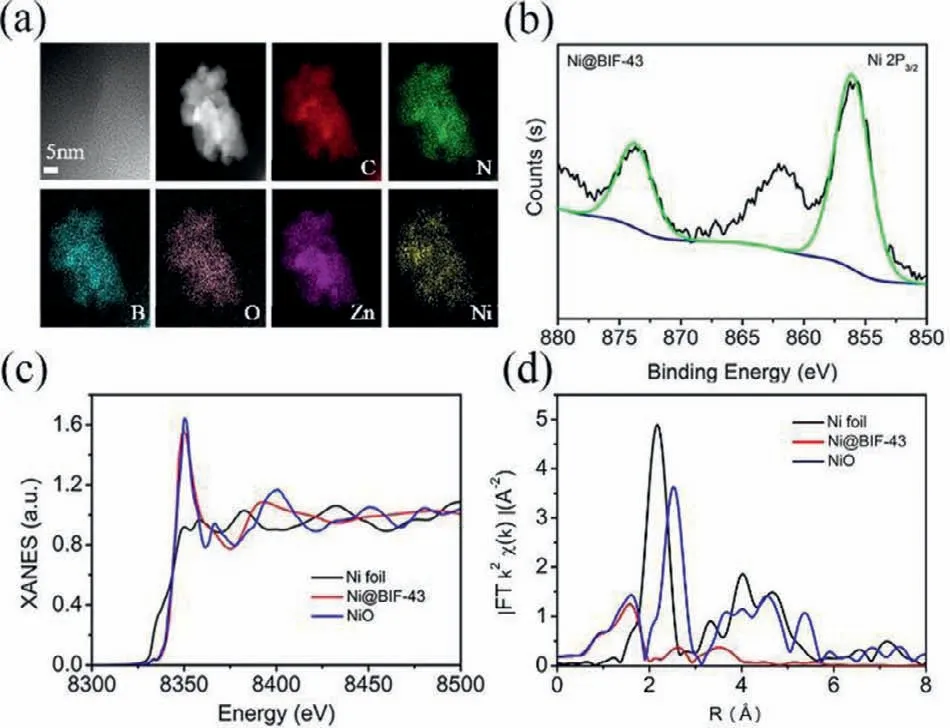
Fig.2.(a) HRTEM images and element mapping of Ni@BIF-43.(b) The high resolution XPS spectrum of Ni 2p for Ni@BIF-43.(c) XANES spectra of Ni@BIF-43 for Ni K-edge.(d) Fourier transformed (FT) k2-weighted χ(k)-function of the EXAFS spectra for Ni K-edge.
Owing to the presence of 2-ATP in the pore wall,the adjacent free-NH2and uncoordinated O-atoms of the 2-ATP could be used to chelate external metal ions in the pores of BIF-43(Fig.1c) [51–53].Therefore,the metalated compounds Ni@BIF-43 were created by immersing the pristine crystals in solutions of Ni2+under air at room temperature.The color of the crystals changed from pale yellow to light green suggesting the adsorption of external metal ions (Fig.S11 in Supporting information).The PXRD (Fig.S12 in Supporting information) demonstrated the retention of BIF-43 crystallinity after loading of Ni ions.Ni@BIF-43 remained porous and showed reduced porosity as demonstrated by N2sorption isotherms (Fig.S8).The BET surface was decreased from 539.8 m2/g for BIF-43 to 275.1 m2/g for Ni@BIF-43.The coordination of Ni ions with the −NH2/O groups of BIF-43 was further confirmed by Fourier transform infrared (FT-IR) spectra.As shown in Fig.S13 (Supporting information),the peaks at 1612 and 1450 cm−1for Ni@BIF-43 displayed a significantly lower intensity compared to that for BIF-43,which are ascribed to the−NH2and −COO stretch vibrations,strongly supporting the interaction between Ni ions and −NH2/O species [54].Inductively coupled plasma atomic emission spectroscopy (ICP-AES) showed that the Ni2+content was 2.1%.The high resolution transmission electron microscopy (HRTEM) images clearly confirmed that no Ni nanoparticles and metal oxides are formed during the immersion period,shown in Fig.2a.Element mapping revealed uniform distribution of Ni ions throughout entire BIF-43 crystal.To gain further structural details on the chemical status and elemental composition of the samples,we performed X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) measurements.The XPS survey scan clearly showed the presence of C,N,O,B,Zn,Ni in Ni@BIF-43 (Fig.S14 in Supporting information).As shown in Fig.2b,the binding energy of Ni 3p peak centered at 856.10 eV,which was assigned to Ni2+[7].The X-ray absorption near-edge structure (XANES) spectra (Fig.2c) also showed that the absorption edge positions of Ni@BIF-43 is also close to that of NiO,which coincided well with the XPS results.In addition,the Fouriertransformed (FT) extended X-ray absorption fine structure (EXAFS)analysis (Fig.2d) inRspace of Ni showed one main peak at 1.56 ˚A,which is longer than the distance of Ni-N (1.40 ˚A) and slightly shorter than that of Ni-O (1.59 ˚A),likely due to the Ni-O and Ni-N combined contributions.No Ni-Ni bonds at around 2.20 ˚A are observed.Altogether,these results underline the presence of Ni2+ions in an atomically dispersed form,which is coordinated to the−NH2/O species of the 2-ATP ligands in BIF-43.
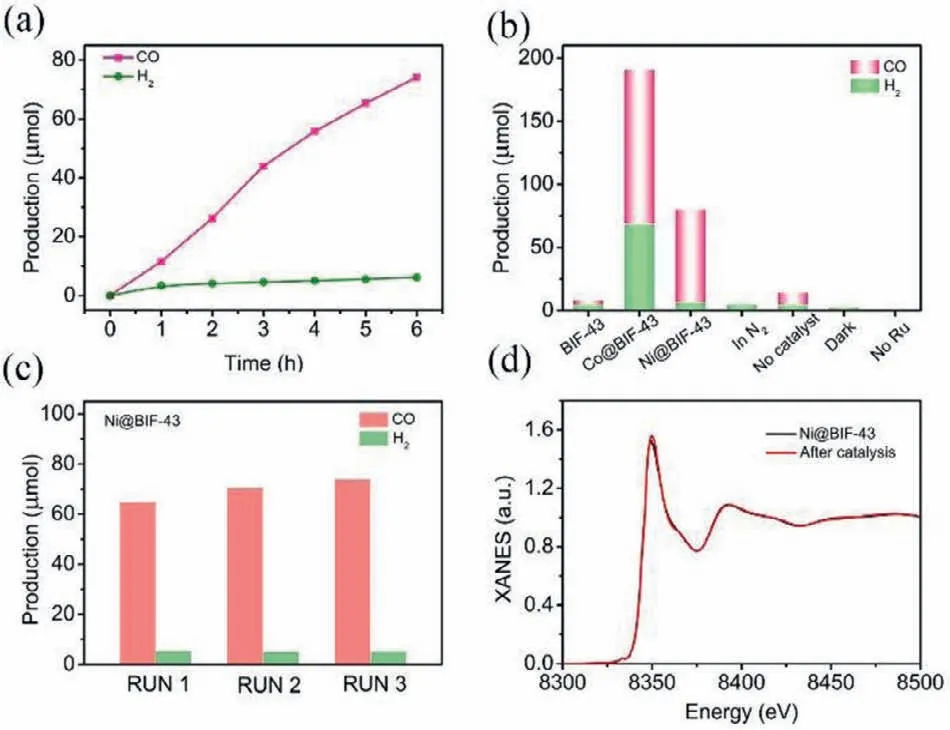
Fig.3.(a) Time course of CO and H2 evolution over Ni@BIF-43.(b) The evolution yield of CO and H2 at different condition.(c) The reproducible photocatalytic activities for Ni@BIF-43.(d) EXAFS of Ni@BIF-43 before and after the photocatalytic CO2 reduction.
Photocatalytic CO2reduction reactions at room temperature have been investigated in CO2-saturated CH3CN/H2O solution with triethanolamine (TEOA) as sacrificial reagent and Ru(bpy)3]Cl2(noted Ru) as photosensitizer under visible light(420<λ <800 nm) irradiation [5,26,44,55].In this reaction system,CO and H2were detected as the main reduction products and no appreciable amounts of other carbonous products were detected from the reaction (Fig.S15 in Supporting information).As shown in Fig.3a,the yields of CO and H2increases almost linearly with irradiation time in a 6 h reaction.The total amounts of CO and H2products from Ni@BIF-43 catalytic system were 66.5 b5~mol (rate: 1109.0 b5~mol g−1h−1) and 7.2 b5~mol (rate:121.1 b5~mol g−1h−1),respectively,giving a high CO selectivity of 90.2%.The apparent quantum efficiency for CO production was measured to be 0.060% at 450 nm (Fig.S26 in Supporting information).It should be noted that the catalytic performance of pristine crystals BIF-43 without anchoring external metal ions was rather poor and nearly undetectable.Therefore,these results demonstrated that the anchored metal ions serve as the catalytic sites for CO2photoreduction [51,52].
Control experiments were carried out to further understand the CO2reduction under different conditions and these results were shown in Fig.3b.No products were detected in the absence of visible light or Ru,suggesting that the CO2reduction is indeed driven by the photoirradiation.When CO2was replaced by N2,only H2can be detected while no CO is observed,implying that the CO derived from conversion of CO2.In the absence of Ni@BIF-43 catalysts,only trace amount of CO and H2were detected,which came
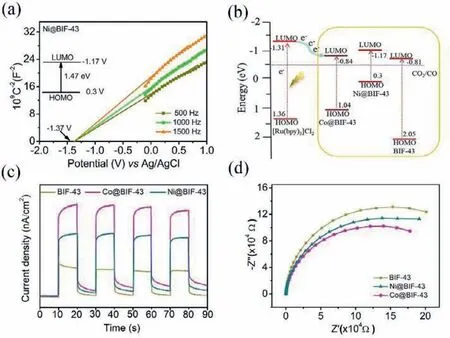
Fig.4.(a) Mott–Schottky plots for Ni@BIF-43 in 0.2 mol/L Na2SO4 aqueous solution.(b) Schematic energy-level diagram showing electron transfer from[Ru(bpy)3]Cl2 to the BIF-43,Co@BIF-43 and Ni@BIF-43.(c) Transient photocurrent response for BIF-43,Co@BIF-43 and Ni@BIF-43 under visible light(420 <λ < 800 nm).(d) Electrochemical impedance spectroscopy plots for BIF-43,Co@BIF-43 and Ni@BIF-43 under visible light (420 <λ < 800 nm).
from the Ru catalysts and were negligible as compared to that with Ni@BIF-43 catalysts.These results confirmed that the CO2reduction reaction was indeed catalyzed by Ni@BIF-43 and driven by light excitation of the Ru photosensitizer.Additionally,Ni@BIF-43 exhibited high durability in cycling tests with reasonably reproducible photocatalytic activities for all three cycles,shown in Fig.3c.XRD analysis (Fig.S16 in Supporting information) evidenced that the crystallinity of the catalysts is maintained.Moreover,XANES (Fig.3d) and EXAFS (Fig.S17 in Supporting information) measurements of Ni have been examined after photocatalytic reaction.No obvious structural evolution has been observed,further reflecting its stability during the CO2photoreduction process.
The well functionalized porous platform of BIF-43 allows us to investigate the significant role of different metal sites in CO2photocatalytic reaction.In comparison,Co with only one d-orbital electron less than Ni has been investigated to construct a counterpart sample of Co@BIF-43.The Co content was 0.52% as determined by ICP-AES analysis.HRTEM,element mapping,photograph and XPS analyses revealed that Co was uniformly distributed throughout the crystal in the form of Co2+ions (Figs.S18-S20 in Supporting information).In the similar photocatalytic condition,Co@BIF-43 displayed a higher photocatalytic activity (2036.0 b5~mol g−1h−1for CO) than that of Ni@BIF-43 (1109.0 b5~mol g−1h−1),however,its selectivity (64.1%) was obviously lower than that of Ni@BIF-43(90.2%) (Fig.3b and Fig.S21 in Supporting information).The apparent quantum efficiency for CO production was measured to be 0.061% at 450 nm (Fig.S27 in Supporting information).In addition,three cycles reactions experiments were conducted to evaluate the stability of the photocatalyst (Fig.S22 in Supporting information).
To elucidate the mechanisms behind the metal sites dependent performance for CO2photoreduction,we studied the whole reaction process systematically.Firstly,the electronic structure of catalyst is one key factor to determine the photocatalytic capability.The UV-vis diffuse reflectance spectra (Figs.S23 and S24 in Supporting information) were conducted to give the bandgap energy of 2.86,1.88 and 1.47 eV for BIF-43,Co@BIF-43 and Ni@BIF-43,respectively.Mott-Schottky measurements were carried out to determine the flat-band position to be −0.81,−0.84 and −1.17 eV for BIF-43,Co@BIF-43 and Ni@BIF-43 (Fig.4a and Fig.S25 in Supporting information),respectively,which are approximately close to the bottom of the conduction band (CB).Combined with the analysis of the UV-vis diffuse reflectance spectra,the energy level diagrams for all three catalysts are obtained (Fig.4b).The CB potential for all the three catalysts were higher than the reduction potential of CO2-to-CO and lower than the lowest unoccupied molecular orbital (LUMO) of Ru (−1.27 eV) [5].These results suggested that these catalysts were thermodynamically capable of receiving the photoexcited electrons from the excited Ru for reducing the adsorbed CO2to CO product.However,although the catalytic activity of Co@BIF-43 was higher than that of Ni@BIF-43,its CB potential was lower than that of Ni@BIF-43.Therefore,energy band positions only endow sufficient driving force to trigger the CO2reduction process but cannot determine the catalytic activity.Furthermore,charge separation efficiency of these catalysts was investigated by the transient photocurrent response and the electrochemical impedance spectroscopy (EIS) under the photocatalytic system condition,shown in Figs.4c and d.Greatly enhanced photocurrent intensity and reduced semicircle arc of Nyquist plots have been observed over Co@BIF-43 and Ni@BIF-43,suggesting the enhanced separation efficiency of the photogenerated electron-hole pairs and faster electron transfer from the excited Ru photosensitizer to the reaction center by anchoring Co/Ni ions.Based on previous works[5,37]and the above results,a conceivable mechanism was proposed.Under visible light irradiation,the photosensitizer (Ru) is promoted to the excited state.This excited state is then oxidatively quenched by the catalyst of M@BIF-43 and transfers electron to the exposed metal active site where CO2molecule is activated and reduced to CO.Finally,Ru returned back to its original state by electron supply from the sacrificial reductant TEOA.Moreover,the highest current intensity and lowest charge-transfer resistance of Co@BIF-43 was consistent with its highest catalytic activity towards CO,which demonstrates that charge separation and transport efficiency may be the key factor for the boosted catalytic reaction rate.Whereas,Ni@BIF-43 shows the higher selectivity of CO over H2,indicating the important selective role of the metal center in the catalytic process.The above results clearly indicate that BIFs modified by transition metal ions can significantly facilitate the charge separation,and vastly improve photocatalytic CO2effi-ciency,and the catalytic performances highly dependent the types of active metal ions.
In summary,an amine-functionalized BIF-43 was reasonably designed and used as the supporter to stabilize active sites Co and Ni,thereby obtaining photocatalysts Ni@BIF-43 and Co@BIF-43.Ni@BIF-43 exhibited a high selectivity of 90% for the CO2-to-CO conversion,while Co@BIF-43 performed a high CO production rate of 2036.0 μmol g−1h−1.The close connection between active sites and adsorption sites,and the enhanced photo-excited electrons transfer together promoted photocatalytic CO2efficiency.Furthermore,the model allows us to realize more definitively the specific effects of different metal-ion species on CO2conversion.The work provided more insights into the design of photocatalysts by anchoring active sitesviafunctional organic ligands as linker.
Declaration of competing interest
The authors declare no conflicts of interests.
Acknowledgments
This work is supported by National Natural Science Foundation of China (Nos.21935010,21773242),National Key Research and Development Program of China (No.2018YFA0208600),and the Strategic Priority Research Program of the Chinese Academy of Sciences (No.XDB20000000).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.10.061.
杂志排行

Chinese Chemical Letters的其它文章
- Photochemical defluorinative functionalization of α-polyfluorinated carbonyls via spin-center shift
- Methods of screening,monitoring and management of cardiac toxicity induced by chemotherapeutics
- Light-guided tumor diagnosis and therapeutics: From nanoclusters to polyoxometalates
- Nanofluidics for sub-single cellular studies:Nascent progress,critical technologies,and future perspectives
- Effective purification of oily wastewater using lignocellulosic biomass:A review
- Recent advances in microchip-based methods for the detection of pathogenic bacteria