Inflammation, microbiome and colorectal cancer disparity in African-Americans: Are there bugs in the genetics?
2022-07-11SamiAhmadHassanAshktorabHassanBrimFranckHousseau
Sami Ahmad, Hassan Ashktorab,Hassan Brim, Franck Housseau
Abstract Dysregulated interactions between host inflammation and gut microbiota over the course of life increase the risk of colorectal cancer (CRC). While environmental factors and socio-economic realities of race remain predominant contributors to CRC disparities in African-Americans (AAs), this review focuses on the biological mediators of CRC disparity, namely the under-appreciated influence of inherited ancestral genetic regulation on mucosal innate immunity and its interaction with the microbiome. There remains a poor understanding of mechanisms linking immune-related genetic polymorphisms and microbiome diversity that could influence chronic inflammation and exacerbate CRC disparities in AAs. A better understanding of the relationship between host genetics, bacteria, and CRC pathogenesis will improve the prediction of cancer risk across race/ethnicity groups overall.
Key Words: Inflammation; African-American; Population-specific genome wide association studies; Minorities health; Microbiome; Colorectal cancer
INTRODUCTION
Colorectal cancer (CRC) is the second leading cause of death amongst cancer patients, an estimated 53000 of whom will die in the United States from CRC in 2021[1]. Strikingly, although total CRC mortality has decreased over the last two decades, particularly in older individuals (age 64 +), CRC incidence has increased in individuals under 50[1]. Recent studies showed early-onset CRC patients were more likely to be African-Americans (AAs), who bear the highest CRC incidence rate between 20-year-old and 44-year-old (7.9/100000) as compared to Caucasian Americans (CAs) (6.7/100000) and Asian-Pacific Islanders (6.3/100000)[2]. AAs commonly display more aggressive types of CRC as well,and are generally diagnosed at more advanced stages of the disease, exhibiting survival rates 7% below those of CAs (58%vs65% 5-year survival)[3]. Such statistics must be interpreted cautiously, since the noted increase of early-onset CRC may result from recently recommended and adopted early colonoscopy (40-45 years old) screening campaigns. Nevertheless, there are multiple proposed influences on CRC disparities in AAs, including differences in health care and treatment access,comorbidities and tumor characteristics[4-8]. Socioeconomic status (SES) also weighs heavily on the late diagnoses and prevention campaign efficacy observed in AA populations[2,6,9]. Undoubtedly the source of CRC disparity is multifactorial, and a layered perspective is imperative to address the alarming rise of early onset CRC, an otherwise preventable disease when detected early.
Herein, we aim to elucidate novel biological factors that may also contribute to the AA disparities in CRC mortality. Specifically, in addition to the genetic influence on CRC pathogenesis, an accumulating body of evidence connects CRC to dysregulated interactions between mucosal innate immunity and the microbiome[10]. Indeed, sustained inflammation promoted by chronic colorectal dysbiosis is an established driver of CRC pathogenesis[11]. Related to this notion is a recent gut microbiome profiling study that found that, in addition to diminished overall species diversity, pro-inflammatoryFusobacterium nucleatum (F. nucleatum) andEnterobacterspecies were significantly more abundant in AA CRC patients as compared to a CA cohort[12]. The presence ofF. nucleatumhas also been linked to inflammation-associated microsatellite alterations found more prevalently in AA rectal tumors, a finding linked to worsened CRC prognosis[13-15]. Furthermore, genetic landscape and microbiome composition have been shown to influence the occurrence of proximal colorectal tumors[16], which are more difficult to detect and are diagnosed nearly four times more often in AA than in CA CRC patients[17]. Nearly 80% of sessile serrated polyps are found in the proximal colon, a phenomenon associated with microbial biofilms andFusobacteria, plus the frequency ofBRAFmutations, CpG island hypermethylation phenotype, and microsatellite instability that increases from the distal to the proximal region[16,18-22].Altogether, there is emerging research on the genetically tuned relationships between mucosal innate immunity, the microbiome, and disparate CRC development, but a functional understanding of how said relationships impact CRC pathology remains incomplete[23]. Additionally, despite the evidence that mucosal innate immunity and the microbiome are intimately connected, this review highlights how minority health research currently evaluates their contribution to CRC risk in a largely separate fashion[24].
Accordingly, we propose an integrated concept whereby a differential mucosal inflammatory response to gut microbiota, influenced by host genetic ancestry, represents an underappreciated factor affecting population susceptibility to CRC. In support of this concept, a recent study found that the most differentially expressed genes (DEGs) between AA and CA CRC tumors were related to the regulation of inflammatory immunity[25]. More broadly, transcriptional regulation of inflammation was determined the most distinguishing DNA variation between African and European genetic ancestries[26].Another study demonstrated that African genetic ancestry and level of African admixture (mixture with ancestral African genetic lineage) predicted a stronger inflammatory transcriptional response in macrophages infected with bacterial pathogens[27]. From this framework, one could theorize that in a CRC setting, the same bacterium or a community of bacteria may induce a differentially deleterious inflammatory response based on a patient’s immune-related genetic background. Positive ancestral selection of anti-pathogenic immunity providing survival benefit in endemic areas may, in a modern“westernized life style” context, exacerbate localized inflammation in the tumor micro-environment and, when compounded by SES and environmental factors, accelerate CRC progression into the more aggressive form seen in AA patients, whose self-reported race/ethnicity correlates with an elevated African admixture[28,29] (Figure 1).
Given that SES and a variety of environmental factors associated with CRC pathogenesis and disparities are commonly discussed elsewhere[2,9], we are limiting the scope of the present review to the nascent literature associating CRC first to genetic polymorphisms related to innate immunity,second to those related to the microbiome, and finally explore how they may conjointly contribute to CRC disparities in the AA population. We also emphasize that, by considering admixture and genetic ancestry rather than self-reported race, population-specific risk studies including microbiome genomewide association studies (GWAS) can more accurately capture human genetic diversity, thereby increasing the likelihood of identifying clinically relevant CRC risk factors associated with African ancestry[30-34]. Furthermore, polymorphisms related to innate immunity as well as the microbiome and contributing to links between CRC risk and African ancestry may have been left undiscovered by the longstanding genetic homogeneity of genomic research cohorts, a problem we discuss in our closing remark. Although technically challenging, expanding GWAS to more diverse “multi-ancestry” cohorts will reveal novel linkages between the microbiome, inflammation, and CRC risk that can build predictive polygenic risk scores adaptable to an equally diverse patient base[35,36]. Crucially, functional evaluation of CRC risk variants associated with African ancestry may offer insights into the trend of aggressive, earlier onset CRC in AA patients, paving the way towards personalized prevention and precision medicine.
MUCOSAL INFLAMMATION, MICROBIOME, AND CRC
To better appreciate how host genetics may impact CRC risk between populations of different ancestral origins by modulating innate immunity or the microbiome, we will first highlight how mucosal inflammation and the gut microbiome interact to affect CRC pathogenesis. The human gut contains up to 1013bacteria that play critical roles in immune, metabolic, cardiovascular, and neurological development[37]. The composition and functions of this bacterial community (microbiota) and its associated genome(microbiome) are highly dynamic and influenced by both environmental factors and host genetic background to maintain immunological and metabolic functionality[38]. Meanwhile, a tightly regulated physical separation between the immune system and commensal bacteria is necessary to limit a chronic inflammatory response to the microbiota[39,40]. The integrity of the intestinal barrier and its epithelium are therefore essential elements of healthy host-microbiota mutualism[40]. To establish a “demilitarized zone” and keep microbes at bay, the epithelium uses different mechanisms including tight junctions between epithelial cells, protective mucus production, and the expression of a complex arsenal of innate receptors that trigger bactericidal mediator secretion[41]. Nevertheless, a permissible level of contact or bacterial penetrance is necessary to facilitate metabolic exchanges and immunity maturation for homeostatic equilibrium between dense microbial flora and the host[39,40].
In the case of a high-fat diet, the cumulative alteration of bacterial metabolites can disrupt this equilibrium, thereby promoting carcinogenic dysbiosis and mucosal inflammation[42]. Diet is, thus, a critical environmental factor when connecting inflammation and the microbiome to CRC risk, especially when considering CRC disparities in AAs compared to Native Africans[43-45]. However, CRC as impacted by the genetic origins of host inflammatory response remains understudied. Inflammatory bowel disease (IBD), a model of perturbed micro-immune crosstalk and a known influencing factor of CRC etiology, can be a useful departure point for this line of inquiry[46]. In fact, multiple GWAS have linked higher risk of IBD, CRC[47-50], and microbiotic dysbiosis to host genetic variations, but surprisingly little is known about how CRC disparities may be compounded by the genetic regulation of host inflammatory response to gut bacteria[51].
There are, however, documented relationships between the genetic regulation of innate inflammatory immunity, the microbiome, and colon carcinogenesis. For example, adenomatous polyposis coli (Apc)Min/+mice knockout for toll-like receptor (TLR) 4 or its signaling adaptor partner myeloid differentiation primary response 88 demonstrated a decreased number of intestinal polyps[52]. Nucleotide-binding oligomerization domain leucine-rich repeat and pyrin domain containing 6 and nucleotide binding oligomerization domain containing protein 2 (NOD2) knockout mice were shown to develop colon tumors following colitis, and fecal microbiota transplantation from these mice into wild type recipients triggered similar tumorigenesis, which interestingly attributed carcinogenic causality to the microbiota[53,54]. In the case of lipocaline-2 knockout mice,Alistipes spp.commensals thrived and drove proximal colon tumorigenesis[55]. The nature of host genetic events can therefore drive different microbiome shifts impacting CRC and its anatomical pathogenesis (i.e.,distalvsproximal).

Figure 1 Immune-related variant may promote survival to pathogens in ancestral African environment but precipitate cancer in descendent African-Americans. Pathogens associated with endemic African regions (e.g., malaria) are thought to pressure selection for specific immunerelated genetic variants associated with pathogen resistance and survival of Native Africans (left). In the context of westernized diet and lifestyle, this genetic predisposition (represented herein by a single nucleotide variant), when associated with inflammatory regulation and inherited from African ancestors, may lead to altered interactions with bacteria or communities of bacteria of the gut microbiome, thereby precipitating the colon adenoma-carcinoma sequence in African-Americans (right). Higher inflammation associated with lack of exercise, high fat diet, and socio-economic status are thought to be predominant factors driving early colorectal cancer onset in African-Americans via their impact on shaping the gut microbiome and its interactions with the host genetic background. SNV: Single nucleotide variant. Created with Biorender.com.
If inflammation is a mechanism connecting the microbiome and colorectal carcinogenesis, host genetic background, including immune-related single-nucleotide polymorphisms (irSNPs), could differentially regulate such associations based on ancestry. In humans, pattern recognition receptor polymorphisms are associated with both IBD and CRC risk[48]. GWAS have linked genetic loci to increased IBD risk and a variety of risk alleles affect immune response[56], including NOD2 or autophagy-related 16 like 1[57,58]. Such polymorphisms have been associated with microbial dysbiosis and an excessive inflammatory response[59,60]. Namely, Knightset al[59] found that among 474 individuals, NOD2 variants were associated withEnterobacteriaceaefamily enrichment, includingEscherichia coli,a species notably enriched in IBD individuals. Also, Lavoieet al[60] described an increase of interleukin (IL)-17-producing CD4+T (i.e.,Th17) cells in the lamina propria of mice engineered to express the polymorphism T300A (rs2241880) in theAtg16 L1gene. Although Th17-based colitis was associated with an increase ofBacteroides ovatus,T300A did not directly induce the increase ofBacteroides ovatusbut rather induced the increase of IL23p19, an important cytokine for maintaining the Th17 lineage[61]. Th17 cells and their canonical cytokine IL-17 are critical pro-inflammatory contributors to epithelial homeostasis and mucosal immunity by orchestrating anti-bacterial defense and epithelial repair and regeneration as well as regulating barrier permeability by controlling the expression of occludin proteins[62,63]. When dysregulated in a chronic setting, sustained IL-17 production may promote colon tumorigenesis[64,65]. Several studies have now identified IL-23/Th17 pathwayassociated polymorphisms linked to IBD susceptibility and the gut microbiome profile[66-68].Presumably, genetic regulation of Th17-driven inflammation may impact IBD and ensuing CRC riskviathe extent or nature of colonic dysbiosis. In sum, these GWAS suggest that by influencing the extent or nature of colonic dysbiosis, genetic regulation of inflammation represents a risk factors for both IBD and CRC. Next, we review how genetic ancestry contributes to this phenomenon, and how it may exacerbate CRC disparity in AAs.
GEOGRAPHIC AND POPULATION-RELATED INNATE IMMUNITY DISPARITIES THAT MODULATE CRC RISK
The flow of genetic information across time and geography may contribute to current disparities in cancer incidence and progression[69]. Cancer is known to result from an accumulation of somatic genetic and epigenetic alterations that dysregulate the cell cycle but also depends on genetic background and polymorphisms that impact patient risk and predisposition[70,71]. Yet, few GWAS have implicated ancestral genetic variants in cancer predisposition amongst self-identified racial/ethnic groups[72,73]. Many such polymorphisms regulate gene expressionviaepigenetic or post-translational modification mechanisms, which affect noncoding sequences like microRNA (miRNA) binding sites[74,75]. Yet, the biological and clinical significance of most polymorphisms associated with cancer disparities remains unknown[72]. Mechanistic associations between chronic inflammation and carcinogenesis could link ancestral genetic diversity to cancer, whereby immune-related genetic regulatory variants have the potential to differentially modulate CRC across populations[27].
In fact, a large fraction of population-associated polymorphisms impact gene expression related to inflammation and innate immunity, which, being likely essential for surviving life-threatening infections, evolved under stronger selection pressures than other traits[76,77]. For instance, human genome diversificationviaarchaic human genome introgression (i.e.,admixture with Neanderthal genome) is a proposed adaptation of ancestral humans to infectious environments following “out of Africa” migration[77,78]. In non-African populations, Neanderthal-introgressed haplotypes reintroduced a splice variant (rs10774671) of the 2’-5’-oligoadenylate synthetase (OAS)1gene[79]. TheOASlocus on chromosome 12 encodes three genes,OAS1,OAS2andOAS3that play an important role in virus defense. The elevated frequency of the Neanderthal-derived allele at theOASlocus was proposed to be the result of a positive selection in European and East Asian populations. This allele selection has a functional significance since it is associated with the production of a protein variant(OAS1 p46) characterized by higher enzymatic activity and improved resistance to West Nile virus and hepatitis virus C[80]. Therefore, the allele haplotype may provide a survival advantage to infectious agents in the non-African environment and represents an example of variety in baseline inflammation levels that may influence susceptibility to diseases like IBD and CRC in patients with insignificant African ancestry. Selection of genetic variants providing health and survival benefit in endemic areas may represent another means of adaptation and human genome diversification. In a cohort of 158 healthy individuals (distributed as European, Sub-Saharan African, and East-Asian), Barreiroet al[81]found that nucleotide diversity of the TLR family was shown to vary between African populations,suggesting pathogen-specific selection pressures. Specifically, theTLR10/TLR1/TLR6locus showed signs of recent positive selection amongst non-African populations. Furthermore, of all SNPs in this region, a high frequencyTLR1single nucleotide variant (SNV) (non-synonymous T1805G variant) found in Europeans was the most significant population differentiator and was associated with a decrease in agonist-mediated nuclear factor-kappa B activation[81]. Although it is unclear if decreased TLR1-mediated immune response confers a selective advantage, it could potentially modulate otherwise harmful inflammatory responses to pathogens[82,83]. This finding suggested that a finely tuned balance between optimal defenses to pathogens and excessive inflammation may have been critical for evolutionary survival[78].
To investigate how ancestral immunity would impact pathogen response, Nédélecet al[27] studied interactions between macrophages and live bacteria (ListeriaandSalmonella). Amongst the macrophages,they found that many of the DEGs; (30% of the 11914 genes analyzed) between AAs (n= 77) and CAs (n= 91) were involved in the regulation of the innate immunity. These results built off their previous findings that 9% of macrophage DEGs varied according to ancestry-associated regulation and that increased African ancestry could predict a stronger inflammatory response to infection[27]. Performing quantitative trait locus (QTL) analysis, the authors identified SNVs in 14% of DEGs or using alternative splicing between CA and AA individual-derived macrophages (either non-infected or infected withListeriaorSalmonella). A large fraction of DEGs were associated with expression QTL only in infected macrophages. In other words, SNVs in a significant fraction of inflammation-related genes were expressed in infected macrophages according to the level of African ancestry. Interestingly, the same authors also found that these DEGs included susceptibility genes previously reported by GWAS for rheumatoid arthritis, systemic sclerosis, or ulcerative colitis, all related to chronic inflammation and conditions with known AA disparity[78]. The interest of such a study, although performed on macrophagesin vitro, is its illustration of the link between African genetic ancestry and inflammatory response to bacteria, one that could accelerate CRC by aggravating interactions between gut microbiota and the mucosal immune system. Reinforcing this concept is another GWAS that revealed that some of the most differentiating irSNPs between African and European populations were associated with genes regulating nuclear factor-kappa B or chemokine gene clusters[78]. Selected genetic variants may offer protection against infection in endemic regions for native/rural Africans but favor cancer development in descendants bearing the same variants in a western environment, a concept exemplifying the crossroad between host genetics and environmental factors that shapes cancer risk (Figure 1).
Regarding the possibility of a role of associations between irSNP and CRC risk into AA disparity, a recent study by Sanabria-Salaset al[33] studied links between pro-inflammatoryIL1Bhaplotypes and CRC risk in patients from six Colombian cities. The authors associated theIL1BCGTC haplotype with CRC risk exclusively in patients from the coastal regions of Colombia who possessed the highest proportion of admixed African ancestry[33]. The same group has associatedIL1BirSNPs (four SNPs -3737C/-1464G/-511T/-31C) with African ancestry and elevated cancer risk. The CGTC haplotype was most frequently found and highly expressed in AAs, establishing a functional link betweenIL1BirSNP and CRC risk[84]. Further studies, validating the connection between AA CRC patients andIL1Bpolymorphisms, will be required to confirm theIL1BSNP haplotype as a population-associated CRC risk marker; these findings nevertheless showcase a prime example of an exploitable connection between ancestry-related inflammation and cancer risk disparity.
Next, a cancer genomic meta-analysis using 48 GWAS within the National Cancer Institute GAMEON Network (64591 cancer and 74467 control patients) across five common cancer sites (ovarian, lung,breast, colorectal, and prostate) found that genetic variants associated with inflammation and innate immune response were relevant to CRC risk, including SH2B adapter protein 3 (SH2B3) (rs3184504,P=3.32 × 10-5), a negative regulator of growth factors and cytokine-induced signaling (Table 1). Unfortunately, irSNP associations with race/ethnicity and geographic distributions were not evaluated[85]. In contrast, Wanget al[47] demonstrated the merit of accounting for population diversity in their analysis of associations between innate immunity pathways and CRC risk. In two large studies (discovery and validation cohorts) across five distinct ethnic groups (AA, CA, Japanese-American, Latino, and Native Hawaiian), they found that among more than 600 common variants associated with 37 innate immunityrelated genes, a SNV in the second intron of peroxisome proliferator-activated receptor gamma(PPARG) (rs9858822) showed a statistically significant association with CRC in the AA population(Table 1). This variant, rare in other non-AA populations, was not previously reported. Importantly, a frequently reportedPPARGvariant rs1801282, inconsistently associated with CRC in other studies[86-88], was not detected in association with CRC in Wanget al[47]’s study that carefully controlled for population diversity, supporting the value of multi-ancestry SNP association studies. In another metaanalysis of 308 SNPs performed by Montazeriet al[89], 14 SNPs showed credible association with CRC including rs3802842, whose expression was associated with immune infiltration.Curiously, thePPARGrs9858822 polymorphism detected by Wanget al[47] failed as a positive SNP in this meta-analysis, a finding that warrants additional verification. However, such discrepancies between the multi-ethnic study of Wanget al[47] and European study of Montazeriet al[89] may highlight the issue of the underrepresented diversity when seeking genetic cancer risk. Population-associated risk may be missed,and alternatively discovered cancer risk may not be relevant for minorities.
Lastly, functional variants in the 3’-untranslated region (UTR) of inflammatory gene miRNA binding sites (miRSNPs) have been associated with CRC risk[75]. Four miRSNPs in the mannose binding lectin 2(MBL2) gene 3’-UTR have been associated with increased CRC risk in the AA population.MBL2codes for mannose binding lactose protein, a pattern recognition receptor that binds a wide range of pathogenexpressed sugars, leading to their phagocytosis. Although not assessed in this study, the modulation of the interactions between the mucosal inflammation and the microbiome,viaMBL2 expression, could be a mechanistic link between African ancestry and higher CRC risk[74]. irSNPs associated with CRC risk are summarized in Table 1.
Ultimately, linking chronic inflammation risk loci to positive selectionviaresistance to past infectious agents and population displacement should be done with caution, as the merits are still debated[78].Physiological interfacing of the immune system with other biological systems (reproduction and organ development) may also explain positive selection in a manner distinct from past pathogen resistance[78]. Yet, considering the evidence of host innate immunity regulation by population-enriched irSNPS[27], it is reasonable to speculate that the mucosal inflammation associated with commensalism is differentially tuned according to the level of African ancestry and could therefore influence CRC disparities.This view is supported by disparity research in other cancers, such as the finding thatIL10promoter SNPs enriched in AAs are also potential risk factors for prostate cancer development and progression[90]. Interestingly, one suchIL10polymorphism (rs1800871) was associated with Proteobacteria load in the gut microbiome (Table 2), but a connection between rs1800871, proteobacteria, and prostate cancer remains to be established[90]. In breast cancer, Jenkinset al[91] demonstrated how the ancestral selection of immune variants in the African continent can predispose AA women to ancestry-related differences in tumor immunogenicity. Specifically, the status of a “Duffy-null” polymorphism-regulated atypical chemokine receptor 1 (ACKR1) allele linked West African genetic ancestry to tumor immune infiltration. Thus, for breast cancer, duffy antigen receptor for chemokines/ACKR1 polymorphism may serve as a biomarker for precision medicine and immunotherapy in patients bearing significant West African ancestry[91]. This result highlights the potential that ancestry-associated irSNPS have for cancer screening and clinical care when paired with functional analyses and elevating the importance of similar studies for CRC.
HOST GENETICS AND MICROBIOME INTERACTIONS’ CONTRIBUTION TO CRC DISPARITY
While host genetics may impact mucosal inflammation and CRC risk, other factors, including environmental factors such as diet, lifestyle, and antibiotic exposure, undoubtedly influence CRC susceptibility and treatment response by shaping gut microbiome composition[51,92,93]. Notwithstanding such findings, we propose that the predominant reliance of the microbiome on environmental cues in healthy individuals may conceal host genetic contributions (including genetic ancestry and somatic mutations)in disease contexts (e.g.,CRC), driving the microbiome response to environmental fluctuations and defining, at least in part, differential susceptibility to cancer in AAs[94-96]. The numerous immunerelated genetic variants that delineate chronic disease susceptibility between AAs and CAs (previously discussed) may then contribute to differential inflammatory responses to the microbiome dysbiosis and compound existing CRC disparities (Figure 2).
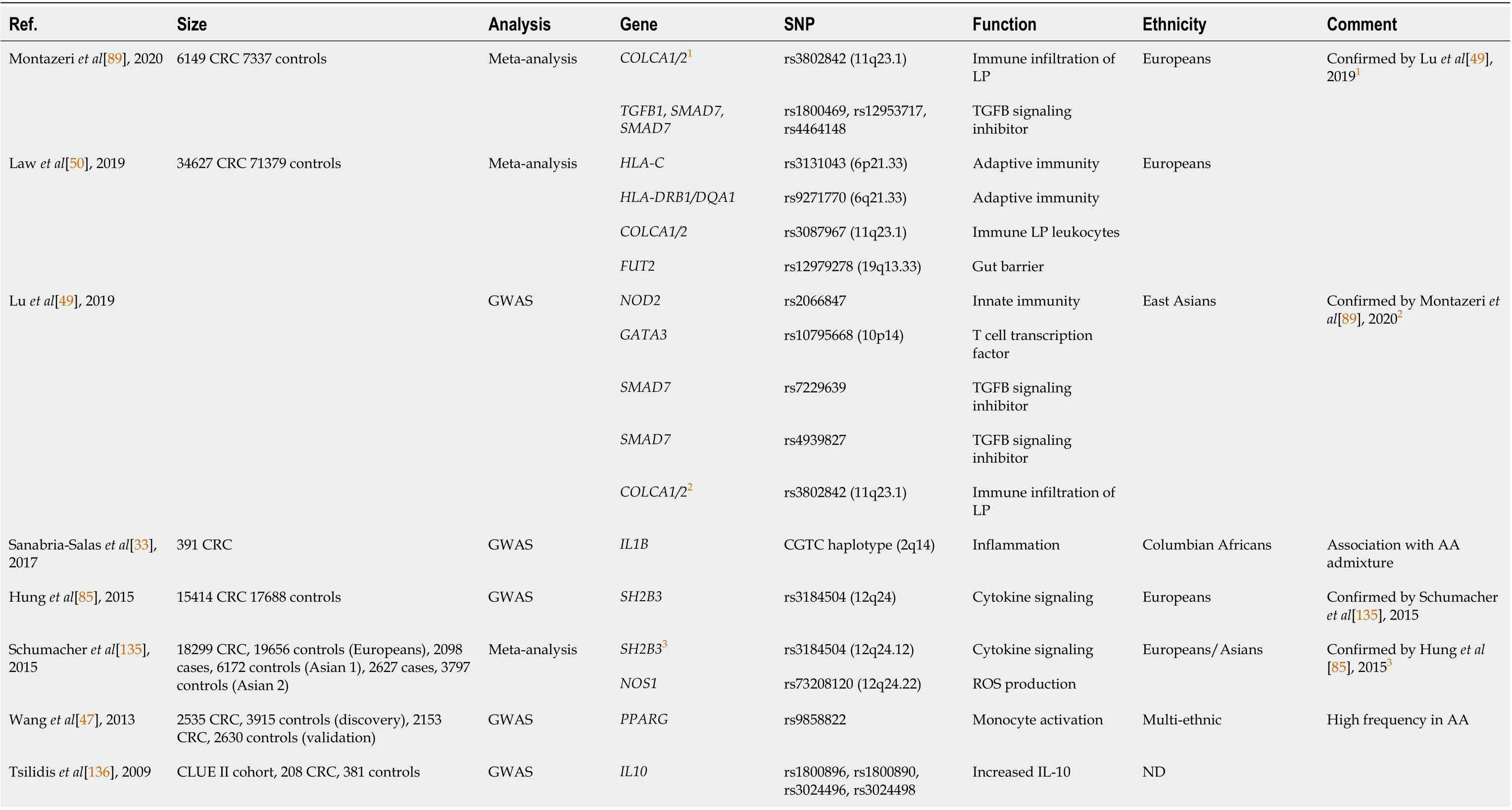
Table 1 Immune-related single nucleotide polymorphism associated with colorectal cancer
From this perspective, multiple metagenomic studies of fecal and mucosal samples have already found compositional and metabolomic differences between CRC and healthy patient microbiome[97-100]. Novel meta-analysis approaches combined these metagenome shotgun datasets across heterogeneous populations to explore relationships between the microbiome and CRC; associations were identified at both bacterial strain and gene levels[101-103]. A common core of 29 bacterial species was enriched in CRC cases, and choline metabolism was established as a reproducible biomarker of the CRC-associated microbiome[101,102]. While such findings are correlative and do not suggest causative links, several smaller studies have highlighted the role of specific bacteria[104-106] and microbial dysbiosis in triggering colorectal carcinogenesis in animal models[107]. However, microbiome GWAS(mGWAS) have not yet identified consistent associations between such carcinogenic bacteria and CRC risk[108]. Unfortunately, because population diversity was systematically underrepresented or not annotated in the metagenome datasets, it remains unknown how these results could translate into CRC risk factors or diagnostic biomarkers for specific racial/ethnic groups.
Meanwhile, several taxonomic and metagenomic studies have revealed an intriguing diversity in microbiome composition across racial/ethnic groups, but mechanistic understandings of associations between bacteria or groups of bacteria and race/ethnicity are sparse[34,109-113]. In particular, diversity of the microbiome is highly diet-driven. A study by O’Keefeet al[44] showed that a 2-wk food swap between AAs (received high fiber, low fat diet) and rural Africans (received low fiber, high fat diet)produced dramatic changes in mucosal biomarkers and a metabolome switch, illustrated by an increase in saccharolytic fermentation and anti-inflammatory butyrogenesis as well as suppression of secondary bile acid synthesis in AAs. In light of these results, it is critical that mGWAS take diet into consideration as a confounding factor, as its impact will inevitably interfere with the genetic/epigenetic influence on CRC risk[114]. Interestingly though, differences between CAs and AAs with respect to the mean alternate Healthy Eating Index (a measure of diet quality[115]) faded when adjusting for SES, implying that diet cannot entirely account for CRC disparities[116]. Multiple other factors besides diet are known to impact the composition and function of the microbiome, including smoking, alcohol consumption, as well as antibiotic exposure or metabolic condition such as diabetes or obesity, which are coincidentally also risk factors for CRC[117-119]. While these aspects of the microbiome biology have been extensively reviewed[120], we are paying much of our attention herein on the role of the host genetics and the ancestral genetic origin on the microbiome diversity and consequently CRC risk between ethnicities.
Interestingly, using the Healthy Life in an Urban Setting cohort and fecal 16S ribosomal RNA gene sequencing of over 2000 individuals, Deschasauxet al[110] found that microbiome diversity between racial/ethnic groups living in the same city was independent of metabolic health and only partially explained by SES, lifestyle, and diet factors. Yet, this finding was not always reproduced in other studies[45,111,121]. Overall, however, there is sufficient evidence to justify additional efforts to clarify if host genetics and population origins are taking part in shaping the microbiome[51,94]. The role of host genetic background has been suggested by mGWAS, which showed that SNPs such as rs4988235,associated with lactase persistence gene, contributed to microbiome composition[45,68,122,123]. A study performed with 416 twin pairs in the United Kingdom identified 26 “heritable” taxa, also suggesting that specific host genetic variants may participate in microbiome composition (Table 2)[124]. Notably,host genetic variants associated with microbiome composition were found to be enriched in immunityrelated pathways[122]. Studies showing associations between metabolic and immune-related polymorphisms and microbiome composition are summarized in Table 2.
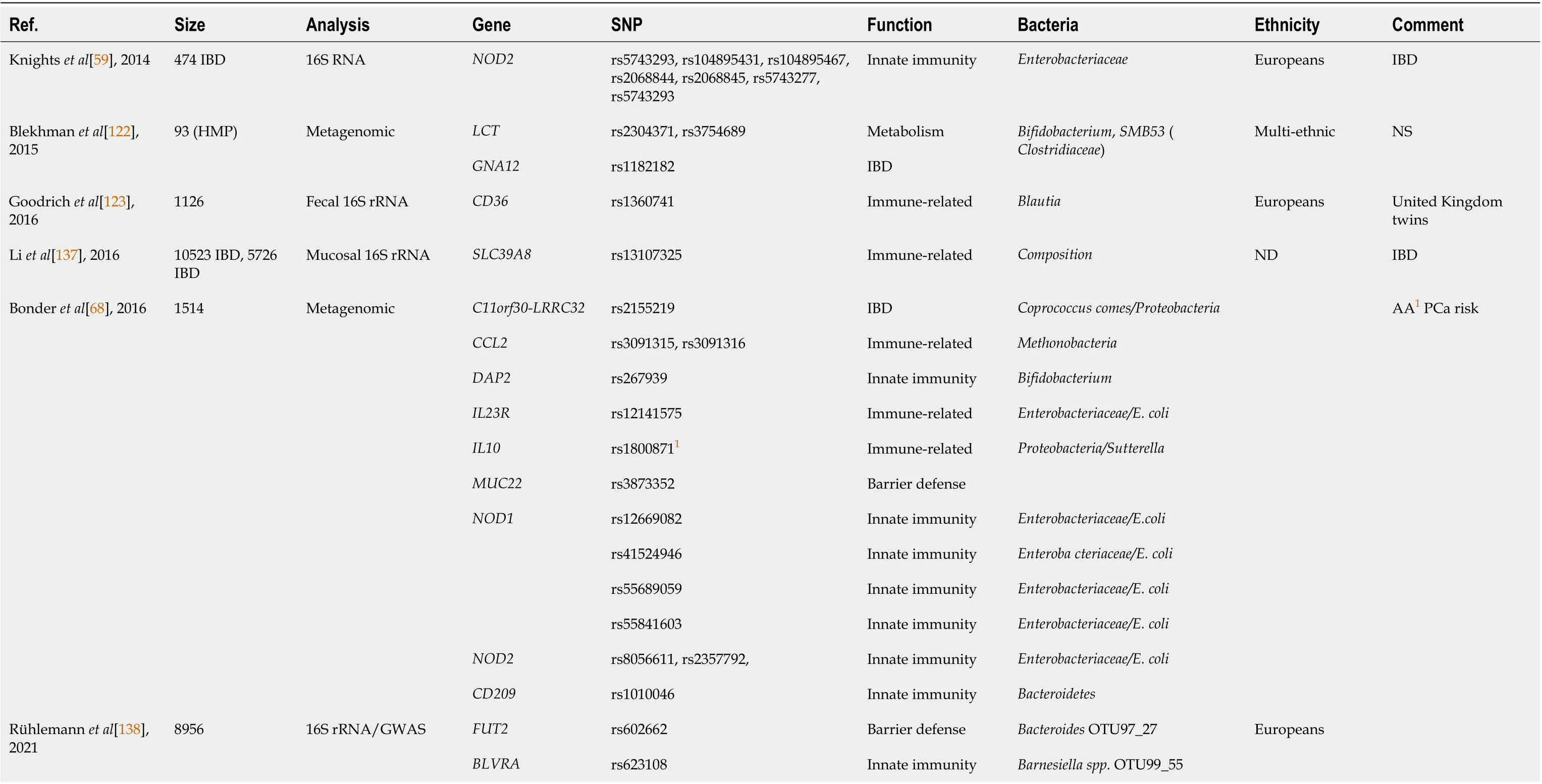
Table 2 Interactions of host genetics (metabolic and immune-related single nucleotide polymorphism) with gut bacteria
Finally, associations between the microbiome and somatic mutations in CRC have been described by Burnset al[95], who suggested that genetic determinants of the host and colon tumor mutations alter microbiome structure. The CRC mutanome could therefore help predict the composition and function of CRC-associated microbiomes and the clinical outcome or the response to therapies. To this end, of intrigue is the recent description by Ashktorabet al[125] and Brimet al[126] of genetic variation in tumor suppressor genes (APC), DNA mismatch repair genes, and other driver mutations (KRASandPIK3C) in AAs with CRC, some of which were novel and not previously described in other populations. Loss of function mutations inAPCwere correlated with changes in 25 different microbial taxa, including an abundance ofFinegoliaorChristensenellaceae. Mutations in the zinc finger protein 717-coding gene were associated with an abundance ofAkkermansiaandVerrucomicrobiaceae,both colitis-associated species.Additionally, the same authors have isolated a novelStreptococcus spp.VT_162 from colon adenoma and CRC lesions in AAs[126]. FecalStreptococcus spp.VT_162 was also confirmed in an advanced adenoma and CRC Chinese/Hong-Kong cohort[126]. An assessment of this bacterium’s relative prevalence in CAs compared to AAs will be necessary. Whether CA genetic background is more restrictive, while AA genetic background is more permissive to this species is not yet established. It may be postulated that,as seen in IBD patients, host genetic background drives the nature of the microbiome and alters the CRC risk posed by procarcinogenic “driver” bacteria[48,51].
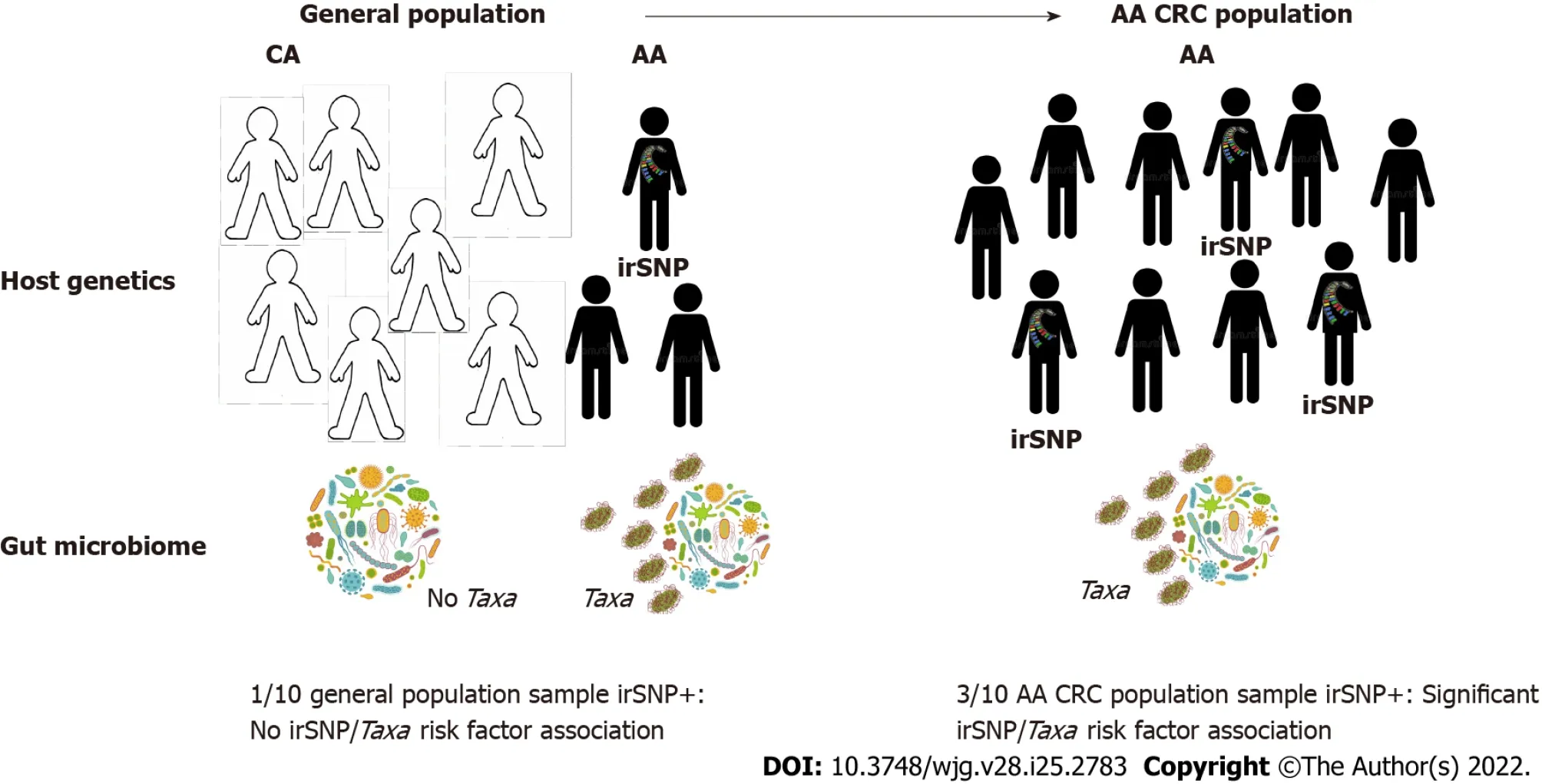
Figure 3 Population-specific colorectal cancer microbiome genome-wide association studies to understand colorectal cancer disparities.Systemic underrepresentation of minorities in microbiome genome-wide association studies (mGWAS) may compromise the identification of microbiome-associated colorectal cancer (CRC) risks in African-Americans (AAs). Left: Associations between AA-enriched immune-related single-nucleotide polymorphisms (irSNPs) (popirSNPs) and gut bacteria (Taxa) may remain undetected in the global population if pop-irSNP and Taxa have too low frequency in the AA population and AAs are underrepresented. Right: If pop-irSNP and pathogenic bacteria are CRC risk factors in AA, they will be enriched in AA CRC patients compared to the general AA population (left), and mGWAS will detect the association between pop-irSNP and Taxa in AA CRC cohort. These features justify population-specific CRC mGWAS to detect additional CRC risk resulting from the interaction between ancestral irSNP and bacteria in minorities. CRC: Colorectal cancer; irSNPs: Immune-related singlenucleotide polymorphisms; mGWAS: Microbiome genome-wide association studies; AA: African-American; CA: Caucasian-American.
Although there is a lack of direct causative links between such bacteria and colon carcinogenesis or growth promotion, this evidence at least signals that CRC disparities in AAs may be related to differential host inflammatory responses to similar bacterial communities (inflammation-driven disparities)and/or the contribution of ancestry-related factors in shaping microbiome diversity (bacteria-driven disparities). The molecular pathological epidemiology (MPE) that aims at uncovering an interactive relationship between environmental features and disease subtypes to understand disease incidence and mortality will provide etiologic and pathogenic insights in CRC disparities[127,128]. However,discoveries made in healthy donors often remain inconsistent from one study to another, perhaps due to multiple confounding environmental factors between heterogeneous cohorts[45,129]. More likely culprits for the inconsistency seen in GWAS that seek to identify the impact of host genetics on microbiome diversity are the homogeneity in sampled cohorts and differences in experimental approaches including stoolvsmucosa sampling, sequencing approaches, and annotations. Future GWAS must address such issues in order to identify associations between genetic background and the microbiome that can reliably be applied to the question of disparities.
CONCLUSION
CRC disparity is far from an exclusively biological phenomenon but rather involves a complex interplay of SES, environmental and genetic components that collectively impact CRC risk and prognostics.Although there is a growing understanding of this complexity, studies examining the influence on CRC pathogenesis from ancestry-specific interactions between host genetics and the commensal microbiome are lacking. Such work is nevertheless urgently needed to appropriately mitigate CRC on a population basis and especially to help address alarming new trends, such as early-onset CRC amongst AAs. To facilitate this line of investigation, we reviewed irSNPs identified in CRC mGWAS and proposed that some of such variants (and others yet to be discovered) may alter microbiome composition and/or differential inflammatory responses to bacteria, thereby impacting CRC risk in a manner associated with genetic ancestry. However, testing the functional significance of such variants will require systematic studies that can incorporate the microbiome, mucosal immunity, and host genetics. A recent investigation by DeStefano Shieldset al[130], although not related to cancer disparity, offers a potential experimental blueprint. Researchers introduced theBRAFV600Emutation to a MinApcΔ716/+murine model of distal colon polyposis, then colonizedBRAFmutant and MinApcΔ716/+mice withEnterotoxigenic Bacteroides fragilis.Distal colon tumorigenesis was observed in MinApcΔ716/+mice following colonization, whereasBRAFV600EMinApcΔ716/+mice developed proximal colon tumors associated with immune signature and microbiome alterations plus sensitivity to anti-programmed death ligand 1[130]. These results suggested that host gene/bacteria interactions may drive CRC risk and pathogenesis, and demonstrate how such interactions can be disentangled mechanistically using experimental models with clinical implications.In the nascent field of precision medicine and multidisciplinary big data integration, the rapidly evolving MPE represents a successful model of integration of pathology, genomics, microbiome,immunology, epidemiology, and social science[127,128]. MPE will considerably improve precision medicine and prevention allowing, among other things, identifying SNPs that may impact microbiome and inflammation and serve as predictive markers. Genetic ancestry, however, remains to be integrated to this model to precisely address CRC disparities at the molecular level. Elucidating the mechanisms by which ancestry-associated variants impact CRC pathogenesis will be much more challenging, but models such as the one described by Lavoieet al[60], who used mice engineered to express the polymorphism T300A (rs2241880) in theAtg16 L1gene known to increase IBD risk in humans, represent a promising approach to identifying mechanisms that can lead to personalized interventions applicable to minorities.
Yet, without addressing biases in genomics science, the ability of GWAS to detect variants of significance for AAs (or other minorities in general) will remain stunted[131]. To date, cancer GWAS have examined cohorts predominantly composed of Caucasian individuals, an homogeneity that limits the appreciation of how genetic ancestry impacts cancer risk[101,102]. Critically, the lack of diverse representation in sampling cohorts results in the increased likelihood that, even if identified, cancerassociated variants may be of limited clinical significance for non-Caucasian populations. While commenting on this issue, Davis[131] recently argued that the current state of genomic medicine is inadequately equipped to confront current oncological trends of disparate incidence and mortality in inclusive fashion and advocated for a persistent push towards the prioritization of patient diversity.Such an agenda is not only more harmonious with the principles of ethical human subjects research but is also scientifically meritable, as studies that account for diversity have already revealed novel genomic data that may improve our understanding of cancer etiology[132]. Therefore, for powered GWAS to detect and discern associations between population-enriched irSNPs, the microbiome, and CRC, proper accounting of population diversity and sufficient cohort size (estimated at > 4000 individuals)[45], or even population-specific CRC studies will be essential (illustrated in Figure 3). Encouragingly, recent methodological frameworks for multi-ancestry cohort GWAS have already yielded ancestry-related cancer variant risk factors[133,134]. Moreover, the recent initiative of the National Cancer Institute:Genetic Association and Mechanism in Oncology (GAME-ON; https://epi.grants.cancer.gov), which regrouped genomic data from more than 33 GWAS across five different cancers (CRC, lung, breast,ovary, and oral), is an example of the benefit of data sharing that will help identifying through metaanalysis data cancer risk loci in understudied populations, especially since 40% of the samples are from African, Asian, and Hispanic backgrounds. However, our integrated concept for CRC genomic research proposes that to represent accurately and capture the contribution of irSNP/microbiome interactions to CRC disparities, such diversified host genomic data must be paired with microbiome data.
In sum, population-related irSNP that regulate mucosal inflammation may modify the microbiota and its interaction with colon epithelium. Alternatively, a population-related SNP not involved in immune regulation may also alter the microbiome and trigger procarcinogenic chronic inflammation.Finally, a combination of two SNPs impacting inflammation and the microbiome with minor effects on CRC pathogenesis may trigger a strong procarcinogenic bacteria/inflammation interaction when combined in an at-risk population (Figure 2). By hunting for and characterizing such genetic factors using the emerging genetic admixture and ancestry paradigms, we believe scientists and clinicians will have a precision tool that enables a clearer understanding of the association between CRC with AA populations and the increasing trend of early onset CRC, ultimately to better mitigate CRC outcome disparities.
FOOTNOTES
Author contributions:Ahmad S wrote the manuscript; Housseau F and Brim H contributed equally to the review’s conception and oversight of the writing, with additional insight from Ashktorab H; and all authors have read and approve the final manuscript.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORCID number:Sami Ahmad 0000-0001-5838-3137; Hassan Ashktorab 0000-0002-4048-4666; Hassan Brim 0000-0001-7055-2298; Franck Housseau 0000-0002-7028-3953.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
杂志排行

World Journal of Gastroenterology的其它文章
- Non-alcoholic fatty liver disease and the impact of genetic, epigenetic and environmental factors in the offspring
- Role of transcribed ultraconserved regions in gastric cancer and therapeutic perspectives
- Multiple roles for cholinergic signaling in pancreatic diseases
- Early gastric cancer presenting as a typical submucosal tumor cured by endoscopic submucosal dissection:A case report
- Correction to “Aberrant methylation of secreted protein acidic and rich in cysteine gene and its significance in gastric cancer”
- Mechanism and therapeutic strategy of hepatic TM6SF2-deficient non-alcoholic fatty liver diseases via in vivo and in vitro experiments