Recent development in selective Tau tracers for PET imaging in the brain
2022-07-09YuyingLiTianqingLiuMengchaoCui
Yuying Li, Tianqing Liu, Mengchao Cui
Key Laboratory of Radiopharmaceuticals, Ministry of Education, Beijing Normal University, Beijing 100875, China
ABSTRACT Abnormal Tau deposition is a crucial pathological hallmark of various neurodegenerative disorders defined as tauopathies, of which Alzheimer’s disease is the most prominent one.To date, a large number of chemical entities with different structures have been developed as Tau imaging tracers for the early diagnosis of tauopathies.Several of them with excellent bio-properties are currently being assessed in clinical trials, and more recently, the TauvidTM ([18F]Flortaucipir, also known as [18F]AV1451 or [18F]T807) as the first Tau tracer was approved by the U.S.Food and Drug Administration in 2020.This review summarized the latest development of Tau tracers and analyzed their chemical structures, with particular attention to the effects of chemical structures on biological properties.In addition, we also discuss the limitations of current Tau imaging tracers, issues that need attention in the development of new tracers, and possible future directions.
Keywords:Tau PET imaging Tauopathies Alzheimer’s disease Selectivity
1.Introduction
As a member of microtubule-associated proteins family,tubulin-associated unit (Tau) highly enriches within the axon of neurons, mainly interacting with the microtubules to regulate their stability and polymerization [1,2].In humans, Tau is a family of six isoforms generated from the alternative splicing of exons 2,3, and 10 of the microtubule-associated protein Tau gene.The exon 10 encodes the second repeat of microtubule-binding domain, and its exclusion and inclusion cause the presence of either three repeats (3R) or four repeats (4R) in sequence [3].Approximately equal amounts of 3R and 4R isoforms are present in the adult brain.Tau is subjected to many post-translational modifications, and phosphorylation is the most common one.The abnormal hyperphosphorylation of Tau decreases its binding to the microtubule, and the detached Tau preferred to self-assemble into insoluble filaments in the neurons and glial cells, which are regarded as a crucial pathological hallmark of tauopathies.The biophysical studies have revealed that these filaments possessed multiple ultra-structures, including paired helical filaments (PHFs), straight filaments (SFs), randomly coiled filaments (RCFs), and twisted filaments (TFs) [4].
Tauopathies are defined as a class of heterogeneous diseases.The spatial distribution and ultra-structure of Tau deposits are different for each tauopathy and are strongly related to the clinical phenotype [5–9].According to the main types of Tau deposits, tauopathies are categorized into mixed 3R/4R, 3R and 4R tauopathies (Fig.1).The mixed 3R/4R Tau isoforms exist in Alzheimer’s disease (AD), chronic traumatic encephalopathy (CTE),primary age-related tauopathy (PART), and Down syndrome (DS).In Pick’s disease (PiD), the Tau deposition is mainly composed of 3R isoforms.On the contrary, accumulation of 4R Tau is characteristic of progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), age-related Tau astrogliopathy (ARTAG), and globular glial tauopathy (GGT).
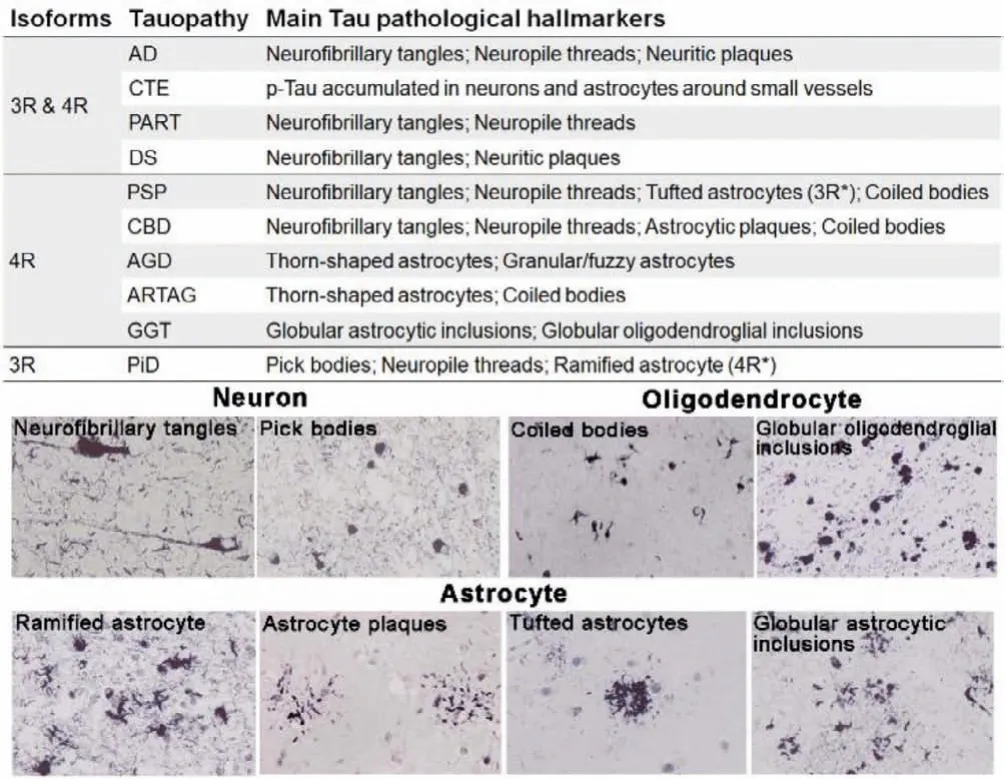
Fig.1.Classification and main pathological hallmarks of the tauopathies.Several typic neuronal and glial Tau pathological morphologies (AT8 antibody).Reproduced with permission [5].Copyright 2014, Oxford University Press.
Furthermore, the affected cell types can vary between diseases[5,7,10,11].In AD and PiD, Tau depositions mainly occur in neurons, presented as neurofibrillary tangles (NFTs) and Pick’s bodies, respectively; meanwhile, PSP, CBD, AGD, and GGT feature as Tau pathologies in glial cells with different morphology.The astrocytes inclusions have complex characteristic structures: tufted astrocytes in PSP, astrocytic plaques in CBD, ramified astrocytes in PiD, thorn-shaped astrocytes and granular/fuzzy astrocytes in ARTAG, and globular astrocytic inclusions in GGT.In comparison,the oligodendroglial inclusions generally appear as coiled bodies.
All these pathological features were observed and studied in postmortem brain tissues.The relationship between the deposited regions and types of Tau filaments and disease is far from being revealed.For AD, large-scale studies have shown that Tau deposits in the hippocampus and entorhinal cortex are related to the severity of cognitive decline, which is also in line with our general cognition because the appearance of NFTs in neurons will directly cause damage to nerve function [12–14].Hence, there is an urgent need to detect Tau filaments in the living brain.Compared with other medical imaging techniques, positron emission tomography (PET)is non-invasive and superior in sensitivity since only picomolar concentrations of radiotracer are sufficient to visualize, characterize, and quantify physiological activities at molecular levels [15].Besides, PET could achieve real-time and dynamically fast monitoring.Therefore, Tau-PET imaging will play a profound role in diagnosing Tau-related disease, disease-staging, and therapy monitoring.More importantly, according to the AT(N) research framework [16], Tau imaging in conjunction with Aβimaging will be very helpful in the early detection and differential diagnosis of AD and non-AD dementias.
Over the past two decades, the development of Tau-selective PET tracers has been driven into an active research scene.There are some reviews about the achievements of Tau tracers in clinical research, but few of them focus on the evolution of chemical structures [17–21].In addition, some new Tau tracers have been reported recently.This review mainly updates current achievements,describing the different structural features of various tracers, focusing on chemical structure, biological activity, imaging performance,and clinical applications.We hope this review will provide general guidance and inspiration for developing new Tau-PET tracers.
2.Tau PET tracers
An ideal Tau tracer needs to follow the general prerequisites of any neuroimaging probes [22–24].(1) The tracers should be easily radiolabeled, with the radiolabeling process both quick and efficient (reaction time ≤1 half-life of radioisotopes, radiochemical yield ≥20%, and molar activity>50 GBq/μmol).(2) The tracer must exhibit high binding affinity toward Tau deposits (nanomolar range forKdorKi) and high selectivity over other misfolded proteins such as Aβandα-synuclein.(3) It should be a small(molecular weight ≤600 Da) and lipophilic (logP=2,3.5) molecule that readily penetrates the blood-brain barrier (BBB) and rapidly washed out from the normal brain.In addition, it should avoid being a substrate for efflux transporter, such as P-glycoprotein (P-gp).(4) Low nonspecific binding to white matter and other neuroreceptors is required to obtain a reasonable imaging interpretation.(5)The tracer should not generate BBB permeable radio-metabolites.Given that PET cannot discriminate between the chemical sources of radioactive signals.(6) For18F-labeled tracers,in vivodefluorination should be avoided, as it will lead to bone accumulation,thwarting accurate quantification of target uptake.
Moreover, the intrinsic properties of Tau proteins should also be considered [25–27].(1) The intracellular location of Tau adds an additional barrier of the cell membrane to BBB that the tracer has to cross by passive diffusion.(2) The concentrations of Tau deposits in the brain are usually 5- to 20-fold lower than those of coexisted Aβaggregates.Thus, more exquisite affinity and selectivity are mandatory for Tau probes.(3) Tau aggregates also appear in brainstem white matter of PSP and CBD, and subcortical white matter of AD and PiD.Thus, the non-specific binding should be prevented in these brain regions.(4) Similarities of six isoforms of Tau protein make matters even worse, and a suitable Tau radiotracer needs to recognize and selectively discriminate these Tau isoforms.In conclusion, there are vast challenges on the horizon for Tau imaging, which seem to be more daunting than Aβimaging.
Based on the structural feature, the reported Tau tracers are summarized as following types in this review: naphthalene, benzothiazole, benzimidazole/indole, quinoline/quinoxaline, nitrogenembedded polycyclic aromatic, pyrrolo-pyridine, and naphthyridine scaffold.The detailed bio-properties of the representative tracers are described in Table S1 (Supporting information).
Since Aβand NFTs share a similarβ-sheet secondary structure,the Tau ligands are commonly derived from the Aβtracers at first.Early in 1999, Barrioet al.first described the naphthalene derivative [18F]FDDNP (Fig.2, [18F]1) as the earliest PET radiotracer for detection of both Aβand Tau in AD [28,29].According to the reported data, [18F]1 possessed a moderate affinity towards Tau aggregates (Kd=36.7 nmol/L) with poor selectivity (only 0.15 against Aβ).Due to the high affinity, [18F]1 could monitor the accumulation of biomarkers in AD and differentiate persons with mild cognitive impairment (MCI) from those with AD and those with no cognitive impairment.However, the non-specific retention in basal ganglia makes some interference.Recently, in consideration of the Tau deposition in CTE is distinctively different from AD, which means that the selectivity is not a requirement for imaging.Then[18F]1 was evaluated for detection of CTE in military personals and retired football players [30].Positive experiment results of [18F]1 in brainstem white matter tracts were consistent with models of concussion while distinctively different from the PHF-tau distribution of AD.The PET imaging can distinguish suspected CTE from the control group and AD patients.The longitudinal studies certified that the PET images were consistent with PHF-tau distribution at autopsy.These results indicated the potential of [18F]1 for early diagnosis of CTEin vivo.This application offered an example for Tau tracers to monitor disease progression in tauopathies apart from AD [31].
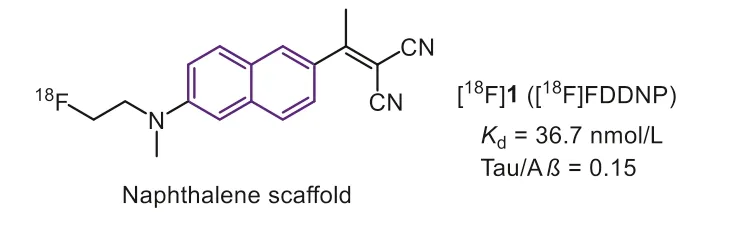
Fig.2.Tau-PET tracers with naphthalene scaffold.
The most common strategy for developing Tau imaging agents is to convert the typicβ-sheet binder, thioflavin T (ThT), to selective Tau tracers.In 2007, Honsonet al.stretched the structure of ThT with a N=N bridge to generate phenyl-diazenyl-benzothiazole(PDB) scaffold as potential Tau probes.However, the selectivity was not robust over Aβ(2-fold) [32].
In 2012, Matsumuraet al.reported a new probe (Fig.3, [18F]2,[18F]FPPDB) by adding a fluoro-pegylation (FPEG) side chain on the PDB scaffold [33].According to the results of inhibition assays, this modification did not improve the selectivity to Tau over Aβ.Profit by this modification, [18F]2 with a low logP(2.05) value displayed good initial brain uptake (4.28 %ID/g at 2 min), but poor washout from the mice brain (2.53 %ID/g at 60 min).Taken together, the unfavorable brain kinetics and poor Tau selectivity impeded its furtherin vivostudy.
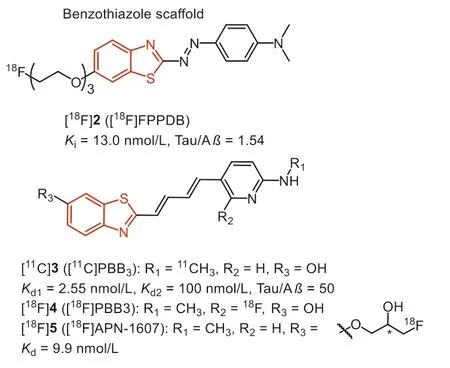
Fig.3.Tau-PET tracers with benzothiazole scaffold.
After screening compounds of various dimensions, the National Institute of Radiological Sciences in Japan, pointing out a specific core length (13–19), might be a significant determinant to get high affinity towards various Tau aggregates [34,35].According to these observations and the known fact that probe with flat and slender configuration could interact with theβ-pleated sheets, they stretched the conjugated backbone of ThT to prepare the phenyl/pyridinyl-butadienyl-benzothiazole or benzothiazoliums(Fig.3, PBBs) scaffold for Tau imaging.Fluorescence staining on brain tissues revealed that PBB1 (clogP=5.44) binds to NFTs and Aβplaques, while labeling of Aβwith compound 3 (PBB3, clogP=3.30) was substantially attenuated, indicating that lipophilicity is another important factor for Tau selectivity.Thein vitrofluorescence staining showed PBB3 could clearly label NFTs and neuropil threads in the brain tissue of AD, PSP, CBD and FTDP-17.Therefore, similar to the design strategy of Pittsburgh compound-B (PiB),compound 3 was labeled with11C, and translated to human PET studies.Phase 0 clinical trials indicate that [11C]3 displayed strong retention in Tau-rich regions of AD patients, in sharp distinction to [11C]PiB, which reflects Aβdeposits.In addition, PET scans using [11C]3 in a patient diagnosed with CBD exhibited tracer retention in the basal ganglia, supporting the detectability of non-AD Tau tauopathies.
However, [11C]3 underwent rapid photo-isomerization when exposed to light, and the isomers showed much less specific binding to Tau fibrils.To prevent this, the radiosynthesis and quality control procedures should be carefully shielded from light.In addition,[11C]3 exhibited fast metabolism with less than 8% of unchanged[11C]3 in human plasma as early as 3 min after injection.The major metabolite, a sulfated compound, is retained in the plasma and gradually enters the brain, making PET quantification unreliable.As the metabolite preserved the backbone of [11C]3, the possibility of this metabolite binding to Tau and other fibrils is required to be further elucidated [36].
Taking advantage of the longer half-life of radionuclide18F(t1/2=110 min), the development of18F-labeled PBBs, [18F]4 (Fig.3,[18F]PBB3) and [18F]5 (Fig.3, [18F]APN-1607) [37,38] for instance,will expand the practical value of commercialization.Consistent with [11C]3, [18F]4 and [18F]5 possessed high affinity to Tau aggregates (Kd<10 nmol/L).Besides, thein vitrofluorescence and autoradiographic labeling of brain sections demonstrated the binding of [18F]4 and [18F]5 to various Tau pathologies in AD, PSP,CBD, PiD, and Tau transgenic mice (PS19 and rTg4510).According to the micro-PET study in rTg4510 mice, [18F]5 underwent faster clearance from the brain than [18F]4, and yielded the highest contrast.In addition, due to the higher brain uptake, [18F]4 and [18F]5 could capture Tau deposits with higher contrast than [11C]3.Notably, the18F-labeled PBBs displayed improved pharmacokinetics,and fewer radio-metabolites in mouse plasma and brain.Besides,[18F]5 displayed minimal off-target in the basal ganglia, thalamus,and choroid plexus, implying the sensitive detection of Tau in PSP and PiD.Although the PBB family suffers from photo-isomerization and a certain degree of off-target effects, [18F]5 was still regarded as the promising Tau tracer for PET, and phase III clinical trial is undergoing in China, Suzhou Xinxu Pharmaceutical Co., Ltd.
In 2005, according to the bio-isosteric replacements strategy,Okamuraet al.explored the benzimidazole derivative (Fig.4, 6,BF-126) with a high affinity for NFTs through screening over 2000 small ligands [39].In neuropathological staining, compound 6 stained NFTs and neuropil threads in the hippocampal section from the AD brain.In contrast, Pick bodies, globose tangles, tufted astrocytes, coiled bodies from PiD, and PSP brain were negatively stained, suggesting it can only interact with NFTs of AD.Additionally, Aβplaques were also stained by 6, although it was much faint compared with thioflavin-S (ThS) staining.It is generally assumed that replacing the benzothiazole with benzimidazole can effectively reduce lipophilicity, making for the selectivity and improving the brain pharmacokinetics.As a result, 6 displayed improved initial brain uptake than 2 (7.2 and 4.28 %ID/g, respectively), and a rapid washout in mice brain (0.16 %ID/g at 30 min).However, further evaluation was hindered by its unsatisfied affinity and selectivity.
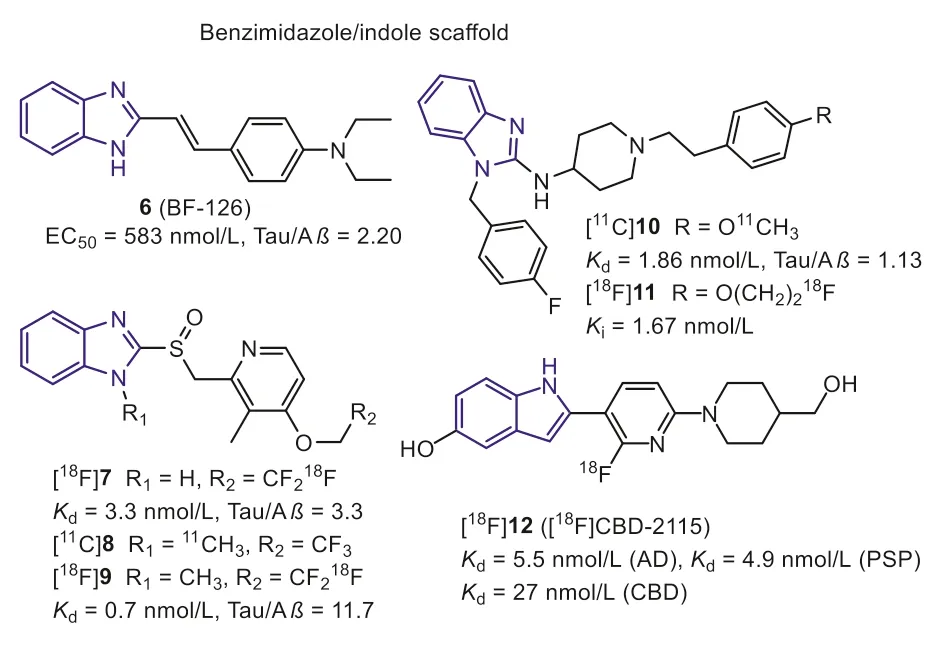
Fig.4.Tau-PET tracers with benzimidazole/indole scaffold.
Subsequently, Rojoet al.found two benzimidazole-based drugs,lansoprazole (Fig.4, 7) and astemizole (Fig.4, 10) for the treatment of allergies and gastrointestinal disorders, hold higher affinity to Tau fibers in 2010 [40].The docking studies indicated that the voluminous moiety on the 2-position of benzimidazole was conducive to interacting with Tau selectively.Compounds 7 and 10 displayed high affinity for synthetic Tau aggregates (Ki=2.5 nmol/L,Kd=1.86 nmol/L, respectively).While they showed lower affinities for AD human brain homogenates (Kd=13.4 nmol/L for 10,Ki=833.3 nmol/L for 7), which may be explained that the synthetic Tau aggregates do not undergo the post-translational modification such as hyperphosphorylation, glycation, and ubiquitination in the native Tau fibers.
In 2012, Xia S.et al.introduced a [11C]methyl group into the imidazole ring of lansoprazole to prepare [11C]8 [41].The affinity of [11C]8 towards Tau aggregates was promoted by this modification (Kd=0.7 nmol/L).In addition, [11C]8 also recognizes 4R-Tau aggregates from PSP patients.However, bio-distribution and micro-PET studies in rodents demonstrated [11C]8 was a substrate for the P-gp, which cannot cross the BBB.Interestingly, [11C]8 showed a high brain uptake in healthy rhesus monkeys (~1600 nCi/cc at 3 min) and decreased to 500 nCi/cc within 40 minutes, suggesting that [11C]8 was not substrate of primate P-gp transporter.Based on these positive results, lansoprazole was regarded as a promising scaffold for developing new Tau tracers.After that, Fawaz M.V.et al.reported two fluorine-labeled lansoprazole (Fig.4, [18F]7 and[18F]9) [42].This study again confirmed that theN-methylation([18F]9) benefits the affinity towards Tau and selectivity against Aβ.The autoradiography study revealed that [18F]7 and [18F]9 specifically labeled Tau on the AD brain sections with a small amount of non-specific binding in white matter.Similar results were expected when imaging nonhuman primates with [18F]9.According to the PET results, [18F]9 holds a comparable brain uptake with [11C]8(peak SUV=2.02 and 1.69, respectively).However, [18F]7 withoutN-methylation displayed very poor brain uptake.The authors speculated that the lansoprazole could easily cyclize to the substrate for the P-gp transporter at lower pH values when theN-methyl was missing.Furthermore, these tracers displayed apparent skull uptake in primates, suggesting lowerin vivostability.Due to these negative results, further studies of lansoprazole derivatives in humans were not carried out.
Similar to the modification of lansoprazole scaffold,11C and18F labeled astemizoles (Fig.4, [11C]10 and [18F]11) were prepared [43].Thein vitrobinding assays demonstrated that the modification on the 4-position of phenylethyl moiety was well tolerated for Tau binding, and allKivalues keep in nanomolar magnitude.Unfortunately, poor brain uptake was observed for [18F]11 inin vivomicro-PET imaging studies using healthy Wistar rats, and there is no improvement after saturation of P-gp.To sum up, the astemizole is also not a preferred lead structure forin vivobrain imaging.
Recently, [18F]12 with the pyridinyl-indole scaffold was reported as a first-in-class 4R-Tau radiotracer [44].In vitrobinding assays showed [3H]12 had a high binding affinity towards Tau aggregates in PSP and CBD brain homogenates, withKdvalues of 5.5 nmol/L,4.9 nmol/L and 27 nmol/L, respectively.Regrettably, these binding results were not confirmed by autoradiography in PSP and CBD brain tissues.In addition, 12 holds two hydroxy groups, sharply decreasing the brain uptake.As anticipated, the PET study pointed out that [18F]16 hardly cross the BBB in rodents and primates[SUVmaxvalue was 0.66 (mouse), 0.5 (rat), and 0.65 (macaca mulatta)], despite it was not substrate of P-gp.
Beyond benzimidazole, Okamuraet al.also discovered that the quinoline derivatives preferred binding to Tau aggregates than Aβ.Inin vitrofluorescence binding assay, 13 (Fig.5, BF-170) and 14(Fig.5, BF-158) exhibited higher binding affinity to Tau fibrils,though the selectivity over Aβwas not ideal (Tauvs.Aβ, 3.6 and 1.7, respectively) [39].Both compounds can visualize NFTs, neuropil threads, and PHF-type neuritis in neuropathological examination using AD brain sections.In addition, an autoradiography study of[11C]14 further confirmed that quinoline scaffold was able to label NFTs in the AD brain.Biodistribution studies of [11C]14 indicated good brain uptake (11.3 %ID/g at 2 min) but a slow washout rate(2.1 %ID/g at 60 min).Although the binding affinity and selectivity were not satisfactory, these results first highlighted the potential of quinoline scaffold for Tau imaging.
After structural optimization of 13 and 14, Fodero-Tavolettiet al.explored a THK family of fluorine-containing quinolines as Tau tracers.Firstly, they prepared fluoro-pegylated quinolines for PET imaging [45].The quantitative affinity showed [18F]15 (Fig.5, [18F]THK523) exhibited 12.4-fold higher affinity toward recombinant Tau fibrils (Kd1=1.67 nmol/L) compared with Aβfibrils(Kd=20.70 nmol/L).Interestingly, the affinity (Kd=86.50 nmol/L)toward AD human brain homogenates was lower than synthetic aggregates.Again, the discrepancy demonstrated the distinction between the artificial aggregates and native proteins.In addition,positive binding of [18F]15 in AD human hippocampal serial sections were co-localized with immunoreactive Tau deposits while failing to highlight Aβplaques, Tau lesions in non-AD tauopathies,orα-synuclein in PD brains.Micro-PET imaging demonstrated significantly higher retention of [18F]15 in the brain of rTg4510 mice compared to their wild-type or Aβtransgenic mice.First-in-human PET studies revealed that the cortical retention pattern of [18F]15 did not correlate with Aβdistribution as assessed by [11C]PiB,whereas following the known histopathological distribution of Tau in AD brains [46].However, high white matter retention of [18F]15 precludes the simple visual interpretation of PET scans and greatly complicates clinical settings.To make matters worse, two independent research groups, Merck Research Laboratories and Siemens Medical Solution, gave challenging opinions on selectivity.Their results indicated that [18F]15 binds to both Aβplaques and NFTs in AD brains [47,48].
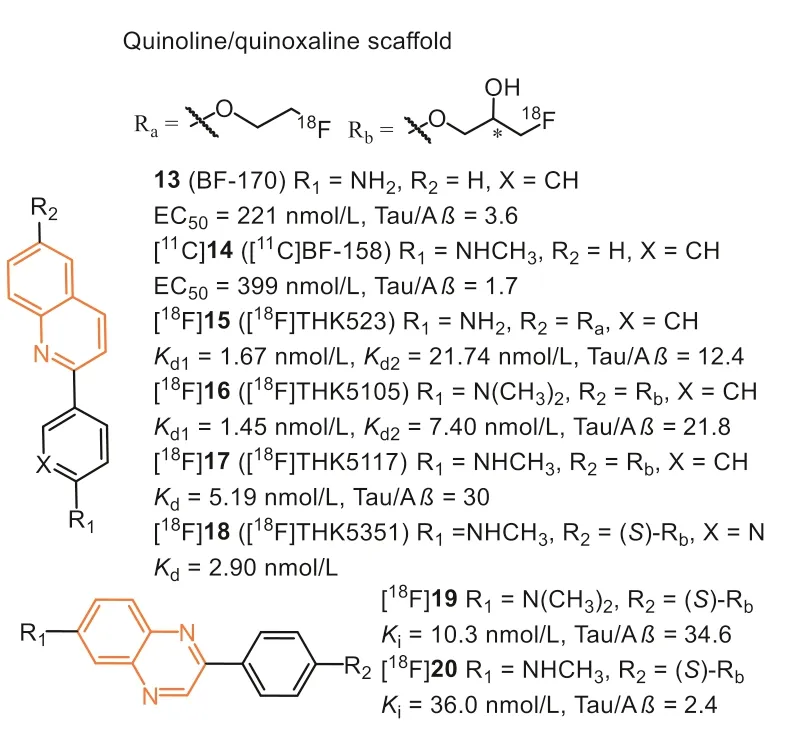
Fig.5.Tau-PET tracers with quinoline/quinoxaline scaffold.
Next, introducing a hydrophilic hydroxyl group on the alkyl chain provided another two quinoline derivatives, [18F]16 (Fig.5,[18F]THK5105) and [18F]17 (Fig.5, [18F]THK5117) [49–52], which exhibited higher affinity (Kd=2.63 and 5.19 nmol/L) than [18F]15.The results suggested that introducing this polar terminus could help the tracers interact with the hydrophilic residues of Tau filaments.Besides, the mono- and di-methylation of the aniline moiety also play a crucial role in enhancing Tau affinity.Autoradiographic study showed that they were preferentially bound to NFTs and neuropil threads co-localized with Gallyas-positive and immunoreactive Tau pathology.Additionally, both of them displayed high initial brain uptake (9.20 and 6.06 %ID/g at 2 min) and rapid brain clearance(1.00 and 0.26 %ID/g at 60 min) in normal mice than [18F]15, which could benefit from the hydrophilic hydroxyl group.The first-inhuman PET studies demonstrated their prominent retention in areas susceptible to Tau deposition in AD patients and distinctly differentiated AD patients from age-matched healthy controls (HC)[53].The retention of [18F]16 agreed with cognitive parameters,hippocampal and whole-brain gray matter volumes, and did not correlate with [11C]PiB retention.However, [18F]16 and [18F]17 displayed non-specific retention in the thalamus, brainstem, and subcortical white matter [49].The reason was mainly supposed to bindβ-sheet structures in myelin, similar to other amyloid tracers.
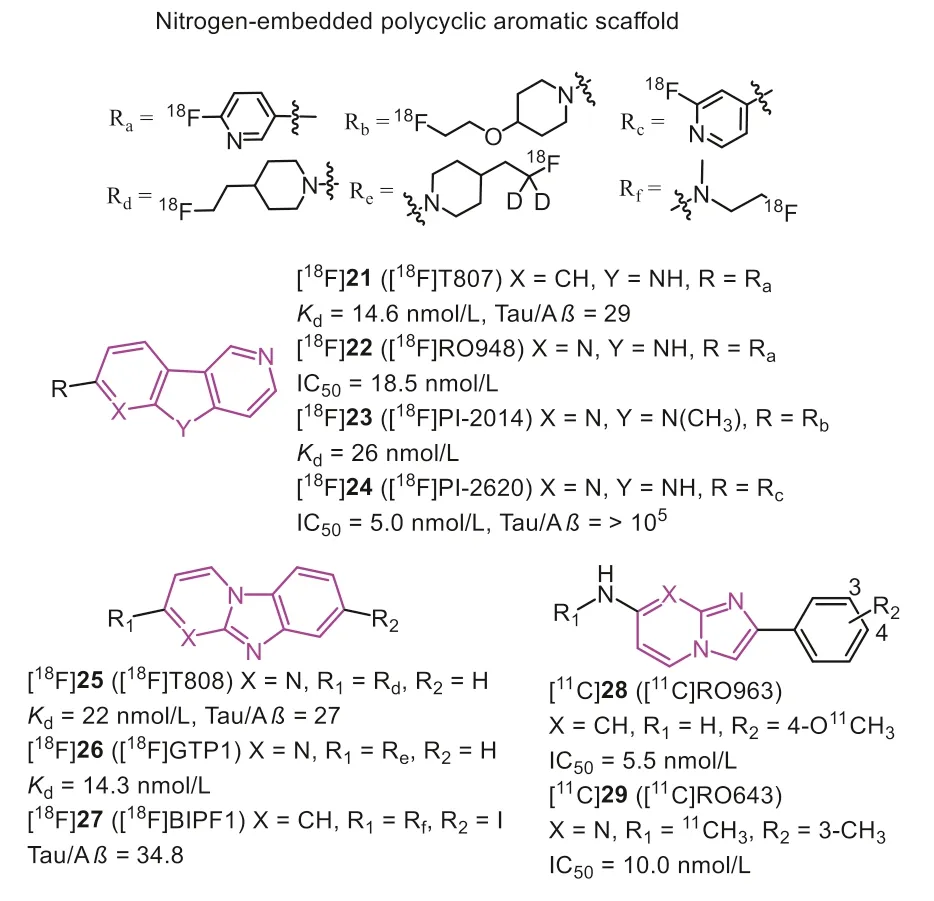
Fig.6.Tau-PET tracers with nitrogen-emmbedded polycylic aromatic scaffold.
Due to the introduction of the hydroxyl group, a chiral center was formed in the case of [18F]16 and [18F]17.Haradaet al.subsequently evaluated their single-enantiomers [54].The results showedS-enantiomers displayed, in general, higher Tau affinity, lower non-specific binding, and faster kinetics than theRenantiomers.Based on this finding, theS-enantiomer ([18F]18,[18F]THK-5351) was prepared for further evaluation.Compared with [18F]17, [18F]18 displayed better Tau selectivity with a higher signal-to-background ratio inin vitroautoradiographic studies.As expected, in human PET studies, [18F]18 showed an excellent ability to differentiate AD patients from HCs.Furthermore, [18F]18 exhibited better washout from subcortical white matter than[18F]17, owing to the replacement of benzene ring with pyridine ring.
However, human PET results suggested that they displayed high non-specific retention in the basal ganglia and other regions with high expression of MAO-B [55,56].The off-target binding was evident in aged patients, restricting it from distinguishing MCI from HCs, even AD.Given the high affinity to MAO-B (IC50=5.2 nmol/L),[18F]18 has been modified as an MAO-B PET tracer for reactive astrocyte imaging [57].
In 2021, Zhouet al.reported a series 2-phenylquinoxaline derivative (Fig.5) for Tau imaging [58].Inspired by the optimization strategy of THK derivatives, the chiral fluoropropanol side chain was introduced at the phenolate position to improvein vivokinetics and increase affinity and selectivity toward NFTs.The binding affinities to Tau and Aβwere determined by using AD brain homogenates competing with [3H]15, [3H]21, and [3H]PiB, respectively.As expected, these tracers bind to NFTs on the THK site instead of the T807 site, and compound 19 exhibited the highest binding affinity (Ki=10.3 nmol/L) and selectivity (34.6-fold over Aβ) towards NFTs, which were confirmed byin vitrofluorescent staining and autoradiography studies.Notably, quinoxalines displayed lower affinity to Aβ, yet again suggesting low lipophilicity might be suitable for binding toβ-sheet in NTFs.Except for this, the brain clearance of [18F]19 was also obviously improved and reached 15.9-fold.Contrary to the findings in THK derivatives,[18F]20 with theN-monomethylamino group showed decreased selectivity and brain kinetics.Recently, Wanget al.reported the firstin-human PET imaging of [18F]19 in AD, and the high contrast images indicated it could effectively distinguish AD from HCs [59].However, similar to the quinoline scaffold, [18F]19 still displayed some off-target binding to MAO in basal ganglia, although the affinity of 19 to MAO-B was 0.33 μmol/L (IC50).
In 2012, Kolb, Zhanget al.discovered that carbazole scaffold could selectively label NFTs on the AD brain slices[60].Through multiple optimizations, [18F]25 (Fig.6, [18F]T808)with a high binding affinity (Kd=22 nmol/L) was reported as a potential Tau probe.[18F]25 displayed excellent brain uptake (6.7 %ID/g at 2.5 min) and rapid washout (2.3 %ID/g at 20 min) in rodents.Thein vivoPET study confirmed that [18F]25 could effectively detect the PHF-Tau in AD patients, and postmortem brain PHF-Tau staining of one AD subject who died two weeks after PET scan was in line with thein vivoPET data of [18F]25.Consistent with the results from the rodents,in vivodefluorination was observed in some cases.
Next, the same group reduced the number of nitrogen on the carbazole and introduced a pyridine ring, eventually identifying [18F]21 (Fig.6, [18F]T807, also known as [18F]AV1451,[18F]Flortaucipir) as improved Tau tracer [48].[18F]21 remained the nanomolar affinity (Kd=14.6 nmol/L) and 25.7-fold selectivity over Aβ.Although the initial brain uptake (4.16 %ID/g at 2 min) was slightly lower than [18F]25, the brain washout was significantly improved in mice (0.62 %ID/g at 30 min).The initial PET scans of [18F]21 in AD, MCI, and HC showed the radiotracer accumulated pattern mirroring the current understanding of Tau deposited patterns described by Braak, where the increased cortical retention was associated with dementia severity based on mini-mental state examination (MMSE) scoring [61].Additionally, [18F]21 exhibited favorable kinetics with rapid delivery into and washout from HC brains.Based on the autoradiography studies, [18F]21 preferred interacting with NFTs and neurites plaques.In other words,it can only recognize the 3R/4R-Tau combinates.Several studies supported this finding,e.g., [18F]21 could clearly label the Tau burden on the CTE brain sections [62].However, clinical studies of PSP patients indicated the uptakes in the corresponding regions were obviously higher than HC and PD, and the uptake pattern was in agreement with neuropathology in postmortem [63].Interestingly,another study showed no significant differences in these regions between PSP and HC [64].Therefore, some researchers concluded that the tracer retentions depended on off-target bindings, mainly on MAO-B and neuromelanin [65].Moreover, a recent PET study indicated [18F]21 could distinguish CBD patients from AD, PSP, and HC, but it also needs more evidence to accurately prove its binding to Tau deposition instead of off-target binding [66].More recently, [18F]21 was reported to have off-target binding in caudate,putamen, pallidum, thalamus, and choroid plexus [67–69].The autoradiographic and relative pathologic studies proved [18F]21 interacted with MAO, dense core Aβplaques, melanin, lipofuscin, and mineralization, which may hamper PET image interpretations.Despite these limitations, [18F]21 showed stronger binding ability and favorable kinetic properties than other first-generation Tau tracers, and U.S.FDA has approved it as the first Tau-PET tracer in 2020 [70].
In 2017, Hoffmann-La Roche Ltd.modified the scaffold of [18F]21 and [18F]25, and reported three ligands (Fig.6, [18F]22 [18f]RO948),[11C]28 (RO963), and [11C]29 ([11C]RO643)) as second-generation Tau-PET tracers [71,72].To reduce the lipophilicity, one more nitrogen atom was added, and finally discovered 22 with high affinity to Tau (IC50=18.5 nmol/L).Additionally, 28 and 29, simplifying the tricyclic aromatic structure to the bicyclic imidazo[1,2-a]pyrimidine/pyridine ring, also possessed delight affinities to NFTs on brain sections from AD.Notably, increasing the nitrogen atoms was not always beneficial for NFTs binding, such as 29 giving a much lower affinity than 28.Autoradiography studies using tritiated ligand verified the excellent specificity and selectivity to NFTs on AD brain sections, and [3H]22 displayed the lowest non-specific binding to white matter.More importantly, no obvious off-target bindings were observed for MAO-A or B.Like [18F]21, these tracers only recognized PHF in AD, but not other Tau deposits in non-AD tauopathies, such as PSP, CBD, and PiD.Next, the brain kinetic was evaluated by PET studies in baboons, [18F]22 displayed the highest initial brain uptake (SUVpeak=2.0) and most rapid washout (11.8)in three tracers.Besides, [18F]22 showed a lower non-specific binding in white matter than [18F]21.The first-in-human studies revealed [18F]22 and [18F]29 could successfully distinguish AD from HC, and [18F]22 showed optimal characteristics in line with preclinical studies (Fig.7) [29,38,54,59,65,71,73–75].Further to this,[18F]22 can also detect Tau accumulation at low concentrations,suggesting that it is possible to monitor changes in Tau pathology over time.
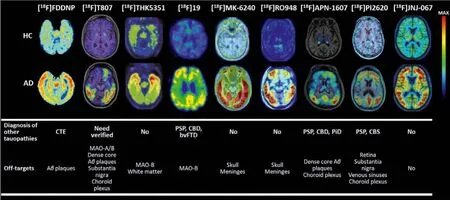
Fig.7.The uptake patterns and binding characteristics of the several representative radiotracers [29,38,54,59,65,71,73–75].Copyright 2002 and 2021, Elsevier; 2022, Wang,Cai, Zhou, Cui and Yao; 2019, American Chemical Society; 2019, 2018, 2021, and 2020, the Society of Nuclear Medicine and Molecular Imaging; and 2020, John Wiley and Sons.
Recently, a head-to-head comparison study showed [18F]22 possessed higher uptake in Tau accumulated areas, such as the entorhinal cortex, and lower retention in white matter [76].Compared to [18F]21, the SUVR of [18F]22 reached equilibrium earlier during the 100 min scanning, which was more suitable for longitudinal assessments.[18F]22 displayed decreased off-target retention in basal ganglia, thalamus, and choroid plexus compared to [18F]21,but there was low-level of age-dependent retention in these regions, implying low but no absent affinity to MAO-A or MAO-B.Additionally, substantia nigra retention in rare cases was observed,which may be caused by the interaction of tracer with abundant neuromelanin.Except for these off-target bindings, the apparent uptake in skull and meninges was reported in some cases.
Recently, based on the structure of [18F]T807 and [18F]RO948,Gabellieriet al.successively identified [18F]23 (Fig.6, [18F]PI-2014)[77] and [18F]24 (Fig.6, [18F]PI-2620) [78,79] as potential Tau tracers for imaging AD and non-AD tauopathies.In vitroautoradiography studies indicated that these tracers could label several Tau types on AD, PSP, CBD, and PiD brain sections, confirmed by AT8 co-localization.However, the major limitation of [18F]23 was the delayed brain uptake (peak at 17 min) and slow brain washout in the rhesus monkey, which contrasted with rodents’results.These disadvantages were likely contributed by non-specific binding in white matter and off-target binding in substantia nigra, basal ganglia, and choroid plexus.Through the structural-activity relationship (SAR) analysis, Krothet al.found that removing theN-methyl group on the tricyclic core would contribute to Tau binding, and decrease off-target binding to MAO-A [80].Nevertheless, the existing NH core might cause low brain uptake and slow washout if a polar group was on the left side.Therefore, fluoropyridine group was a good option, and replacing the 2-fluoropyridin-5-yl substituent with the 2-fluoropyridin-4-yl increased the selectivity over MAO-A.Further to this, the position ofN-atom in the right pyridine ring played a key factor for Tau-binding.In contrast, theN-atom in the left pyridine ring mainly affected the MAO-A binding.For instance, compounds 22 and 24 displayed higher IC50values for MAO-A than 21.Tracer [18F]24 with higher Tau affinity(IC50=5.0 nmo/L), better brain pharmacokinetic, and lower offtarget binding to MAO-A (IC50>1000 nmo/L) was finally identified according to these modifications.The first-in-human study verified that [18F]24 could discriminate AD from HC with no significant retention in the off-target binding regions (e.g., basal ganglia, choroid plexus, and meninges) [81].While a recent study indicated, there are off-target bindings in the retina, substantia nigra,and venous sinuses, and some cases showed low levels of choroid plexus bindings [82].Besides, the mild off-target binding to TDP-43 may be captured in the anterior temporal pole of a semantic variant primary progressive aphasia (svPPA) patient, although it contradictedin vitroresults.But generally, [18F]24 displayed much lower off-target retentions than [18F]21, and owing to the rapid washout from the white matter, [18F]24 was more beneficial for assessing the early stage or minimal changes of Tau pathologies.Chotipanichet al.described that the appropriate imaging window of [18F]24 was 30–40 min post-injection, which is much shorter than other reported tracers (70–90 min) [83].Another study in 7 subjects (including 3 HCs and 4 ADs) pointed out that static [18F]24 PET scans between 45–75 min post-injection displayed high quantification accuracy, large effect size, and low test-retest variability,which could contribute to longitudinal studies [84,85].Moreover,[18F]24 can significantly bind to the globus pallidus internus in the PSP brain, embodying its value in PSP diagnosis [86,87].Except for this, [18F]24 also demonstrated its efficacy in imaging 4RTau isoform from corticobasal syndrome (CBS) [88,89].However, it is also important to point out that the brain clearance of [18F]24 was higher in PSP and CBS than in AD, suggesting a lower binding affinity to 4R than 3R/4R combinations.
In 2019, Sanabria Bohorquezet al.reported [18F]26 (Fig.6,[18F]GTP1) [90], a deuterated analog aiming to reduce thein vivodefluorination of [18F]25.[18F]26 has been proved with nanomole affinity to Tau and high selectivity over Aβby autoradiography studies on AD brain slices, and the results also suggested no offtarget binding, particularly to MAO.According to the PET imaging, the isotopic substitution of hydrogens on the carbon carrying18F indeed decreased the defluorination of the tracer in mice and rhesus, but low levels of uptake were still observed in the skull(14.3 %ID/g at 30–45 min in mice).Fortunately, there was no evidence of defluorination in human PET studies, and [18F]26 exhibited specific cortical retention in AD [91].Furthermore, another longitudinal cognitive performance indicated [18F]26 holds a robust SUVR in cortical gray for prognosing cognitive decline among mild ADs, suggesting [18F]26 PET imaging may serve well for AD progression in the clinic [92].
In 2016, Onoet al.found that removingN-atom on the left ring of [18F]25 would not influence the Tau affinity and reported a series of125I-labeled racers based on the benzimidazopyridine(Fig.6, BIP) scaffold [93,94].The autoradiography studies showed these tracers could label the Tau deposits on AD brain sections rather than Aβplaques.Notably, alkylation on the amino group improved Tau selectivity and brain kinetics.Then, a fluorine atom was attached to the terminus of ethylamino group to produce[18F]27 with excellent Tau selectivity over Aβ(34.8-fold), high initial brain uptake (brain2min=6.22 %ID/g), and acceptable washout(brain60min=2.01 %ID/g) [95].To overcome the poorin vivostability of [18F]27, they explored various substituents at the 7-position of BIP scaffold [96].However, the negative results suggested that 7-position was not crucial for improving affinity and pharmacokinetics.Inspired by the improvement of [18F]21, they investigated whether introducing another nitrogen atom on the fused tricyclic structure will promote affinity and brain kinetics [97].The data implied the brain washout ratio would be improved by adding the nitrogen atom either on the left or right ring of the 6,5,6-tricyclic scaffold.In contrast, transferring the nitrogen from the left ring to the right one, leading to pyridoimidazopyridine (PIP) scaffold, displayed improved Tau selectivity over Aβ, consistent with the findings in [18F]24.Overall, the BIP and PIP scaffolds possessed good binding affinity and selectivity to Tau pathology of AD brain sections, and it deserved further modification to be translated to clinical studies.
In 2016, Merck sharp and Dohme Corp.reported pyrrolo[2,3-c]pyriding structure as the potential backbone for screening Tau PET tracers (Fig.8) [98,99].Throughin vitrocompetitive binding assay using AD brain homogenates, the 3-(1H-pyrrolo[2,3-c]pyridin-1-yl)isoquinoline was identified as the lead optimization(Kivalues were 0.3 and>104nmol/L for NFTs and Aβ, respectively).According to SAR analysis, the 1-(5-methylpyridin-2-yl)-1H-pyrrolo[2,3-c]pyridine was regarded as the minimal pharmacophore.In addition, introducing fluorine atom at the 6- or 8-position of isoquinoline moiety has less effect on the Tau affinity.PET study of [18F]30 in rhesus monkeys displayed apparent retention in white matter due to the undesired high lipophilicity (sfLogD=3.6).The addition of nitrogen atoms was a straightforward strategy to reduce non-specific binding, whereas compound 31 displayed a sharp loss in affinity to NFTs after converting the isoquinoline to the naphthyridine ring.Therefore, they decided to introduce an amino group into the 5-position of isoquinoline, and the final [18F]32 ([18F]MK-6420) exhibited high Tau affinity (Ki=0.36 nmol/L, and selectivity was 105-fold) and better brain pharmacokinetics.In the first-in-human study, [18F]32 displayed high brain penetration (SUV=3–5) and uniformly rapid brain clearance in HCs [100].By contrast, the slower clearance was observed in regions with NFTs deposition in AD, and the spatial binding patterns were confirmed by [18F]25.It was even more pleasing that [18F]36 overcame the off-target binding, which was commonly appeared in the first-generation tracers [74,101].A recent report compared the PET results between [18F]36 and [18F]25 in the same subjects [102].The former displayed much lower offtarget bindings in the striatum and choroid plexus, but the retentions in the meninges were more frequent than [18F]25.The data also indicated [18F]36 possessed a 2-fold higher dynamic range of SUVR, which may be reasonably attributed to the 3- to 10-fold higher affinity, and more suitable metabolism, reference region clearance, and otherin vivofactors.It will be advantageous to diagnose Tau pathology in early-stage and perform longitudinal studies to detect small interval changes.A recentin vivostudy found that [18F]36 provided consistently accurate data to diagnose AD from preclinical to symptomatic stage [103].It should be noticed that [18F]36 can not recognize other Tau isoforms in non-AD tauopathies.Currently, [18F]36 has been undergoing phase III clinical trials in the U.S.
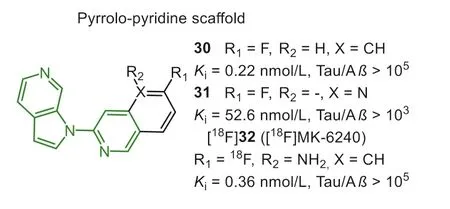
Fig.8.Tau-PET tracers with pyrrolo-pyridine scaffold.
In the same year, Janssen Pharmaceutica N.V.identified thatN-(6-methyl-2-pyridyl)quinoline-2-amine (Fig.9, 33) hold good affinity (Ki=13.9 nmol/L) and acceptable selectivity (10-fold) by a minihigh throughput screening on 4000 compounds [104].They subsequently optimized this lead by strict SAR analysis to achieve more selective Tau-PET tracers.Firstly, they found that the quinoline nitrogen was crucial for Tau selectivity, while the variation of the nitrogen position on the affinity was unclear.The results proved quinoline and isoquinoline derivatives both displayed affinity to Tau aggregates, while compounds with quinazoline scaffold preferred to interact with Aβ.Similar to the findings of quinoline core, pyridine nitrogen was also important for affinity towards Tau, and the tolerated modification was thep-oro-position of quinoline substituent.Interestingly, the pyrimidine scaffold, holding two nitrogen atoms on these two positions, would remarkably decrease the affinity toward Tau.Next, directly introducing aromatic fluorine on the quinoline ring was a suitable strategy for achieving PET imaging.On the one hand, modification on the pyridine ring would remarkably affect the selectivity because the basicity of the pyridyl nitrogen is a key factor in obtaining good Tau affinity.On the other hand, the fluoroalkyl-substituent tends to cause unsatisfactory brain radio-metabolism.Finally, compound [18F]34 (Fig.9, [18F]JNJ-311, 6-flouro-N-(2-methylpridin-4-yl)-1,5-napathyridin-2-amine) had been optimized as the final Tauselective tracer.In comparison with [18F]21, [18F]34 showed higher affinity (Ki=7.9 nmol/L) and more considerable selectivity against Aβ(>540), confirmed by displacement studies with [3H]21 on AD brain sections.However, it should be noted that no specific binding was seen in the frontal cortex on PSP and CBD brain sections,which means [18F]34 was not appropriate for the diagnosis of non-AD tauopathies.Mouse biodistribution studies showed [18F]34 possessed a moderate initial brain uptake (SUV=1.9 at 2 min) similar with [18F]21 (SUV=2.2 at 2 min), but faster brain washout (2–60-brain uptake ratio was 29.6 and 18.3 for [18F]34 and [18F]21, respectively).The similar initial brain uptake and washout ratio were examined in Wistar rats (SUVs=1.5 at 2 min; 0.2 at 60 min) and rhesus monkeys (SUV=1.9 at 1 min and 0.4 at 60 min), according to the animal PET scans.Notably, the gradual bone uptake was only observed in rats but not in mice and rhesus monkey, and this interspecies difference was also seen in [18F]21, even in other reported PET tracers.Moreover, in line within vitrostudy, negative results ofin vivoself-blocking and displacement, suggested that no off-target binding happened in healthy rat’s brains [105].
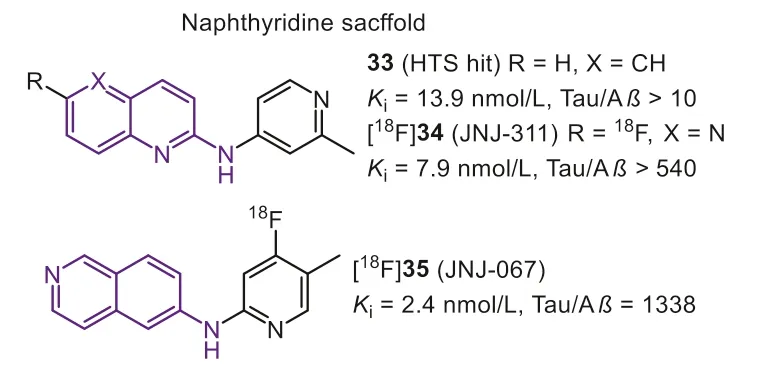
Fig.9.Tau-PET tracers with naphthyridine scaffold.
To further optimize the pharmacokinetic profiles, a similar design strategy for [18F]34 was applied, and then [18F]35 (Fig.9,[18F]JNJ-067) with isoquinoline scaffold was reported [106].Same as previous findings, when the nitrogen was decided on 6-position of isoquinoline moiety, to keep the high affinity and selectivity,the pyridyl nitrogen should be located at the 2-position, and the methyl should be shifted into the 4-position.[18F]35 possessed an excellent binding affinity (Kd=2.4 nmol/L) and selectivity (1338-fold over Aβ) that was twice higher than [18F]34.Autoradiography studies in AD brain sections confirmed the high selectivity and low non-specific binding in white matter.Like [18F]34, [18F]35 showed no off-target binding in rodents and primates.More importantly, this modification positively improved the initial brain uptake, which holds a considerable uptake in rats (peak SUV is 2.4)and rhesus monkeys (peak SUV is 5.4).Based on the above promising results, human studies of [18F]35 were subsequently completed[75,107].[18F]35 clearly labeled NFTs in AD patients with low accumulation in HCs.Whereas the quantitative analysis indicated no significant difference in any ROIs for MCIs or PSPs relative to HCs,suggesting [18F]35 cannot diagnose early-stage AD and other non-AD tauopathies.
3.Discussion
In the past decades, the development of Tau-PET tracers has been regarded as one of the hot spots in radiopharmaceuticals and neuroscience.Many tracers with different molecular entities were discovered, and several have been achieved in preclinical or clinical studies that yielded encouraging data.Especially in 2020, U.S.FDA approved the first Tau-PET tracer, [18F]21 ([18F]Flortaucipir).At the same time, there are still many issues that await further exploration and discussion.
Firstly, it should be noted that the binding affinities of the reported tracers are not comparable due to different measuring methods and materials.As described above, heparin-induced synthetic Tau polymers, postmortem AD human brain homogenates,and AD brain sections are routinely used inin vitrocompetitive inhibition and saturation binding assays to determine tracers’binding characteristics.However, the affinities measured by synthetic Tau aggregates and native Tau fibrils for the same tracer were sometimes quite different, possibly due to the lack of posttranslational modifications in the former, in contrast to Aβ.It is well known that phosphorylation, ubiquitylation, glycosylation,nitration, and acetylation are closely involved in Tau misfolding,and together constitute the binding surface of theβ-sheet domain[108].Therefore, the binding of the tracers to the synthetic Tau aggregates would lack the interaction brought about by these modifications.
In addition, the affinity data measured by different types of competing ligands can only be defined as relative values in most cases.It is generally known that there are multiple binding sites in the Tau fibrils.Caiet al.found two different binding sites on native NFTs for [18F]14 and [18F]21, which were confirmed to be different from PiB [109].Similar chemical structures share the same binding site, and for instance, the quinoxaline derivatives compete well with the THK series.Unlike other tracers, compound 7 has at least two NFTs-binding sites (Kd1=2.55 nmol/L andKd1=100 nmol/L), one of which is identical to the specific site of 21 according to the blocking experiment [110].In a recent study, Muruganet al.also confirmed that compound 7 acts on multiple binding sites of NFTs, with the most potent binding site different from other tracers.With the help of computer-aided molecular docking studies, four high-affinities binding sites, including three core sites(S1, S2 and S3) and one surface site (S2) were revealed in the Tau protofilaments.This study also revealed that the tracers interact with Tau mainly through the hydrophobic bridge within the surrounding residues, and at least one preferential binding site [111].For example, 18 binds strongly to S3 and S1 sites, with S3 being preferred.Collectively, disparate evaluation standards and measurement methods of bind affinity would further complicate tracer screening.
Next, it is well known that many misfolded proteins, including Tau, Aβ,α-synuclein, and TDP-43, may coexist in the brain,especially since Aβhas a higher concentration in the AD brain.Therefore, Tau tracers need to have a more pronounced binding affinity and selectivity to Tau filaments.However, most of the first- and second-generation Tau tracers were discovered and optimized through high throughput screening.The mechanism of the tracer binds to Tau fibrils is not fully understood.Although limited optimization strategies provided some clues, for example, better Tau specificity could be achieved by extending tracer’s molecular length or reducing lipophilicity.However, the data of [18F]32 pointed out that adding additional hydrophilic nitrogen atoms may disrupt the brain pharmacokinetics of tracer.In conclusion, these strategies do not provide perfect reference rules for the rational design of tracers.
Moreover, these optimizations aim to reduce off-target binding,especially MAO.The results demonstrated that second-generation Tau tracers significantly attenuated off-target binding to MAO.PET images showed no significant difference between AD and HC in basal ganglia.However, further PET studies and histopathological experiments have displayed that these probes still have abnormal retention in other non-target regions, such as the skull, choroid plexus, retina, substantia nigra, and venous sinuses.During human PET scans of [18F]22, [18F]24, and [18F]32, skull uptake was observed in some cases, but whether this comes from defluorination is unclear.[18F]21 and [18F]5 exhibit off-target binding in the choroid plexus, which is adjacent to the hippocampus and thus may interfere with the quantification of Tau burden in this critical pathological region [112].These off-target bindings are attributed to melanin, lipofuscin, iron, and calcium accumulated.However,more research should focus on the binding mechanism to reduce these off-target bindings.
Finally, whether the reported Tau tracers can diagnose other tauopathies have received extensive attention.As described in the introduction section, Tau pathologies are heterogeneous among different Tau lesions.At present, few tracers could detect Tau depositions in non-AD tauopathies, particularly with single 4R- or 3R-Tau isoforms.It should be emphasized that [18F]5 with a longer core length could recognize almost all Tau isoforms and successfully diagnose PSP, CBD, and PiD.[18F]12, with a similar elongated structure, also preferentially interacts with 4R-Tau isoforms.Besides,the multiple preferred binding sites for [18F]5 may also contribute to recognizing distinct Tau filaments in different tauopathies.It is worth mentioning that, in recent years, the structures of Tau filaments from AD, PiD, CTE, CBD, PSP, GGT, and AGD have been elucidated successively by cryo-EM studies [113–117].These studies have shown that the Tau filaments exhibit different structures in various diseases with minor differences in the same Tau subtype.More information about the Tau aggregation and folding will help us better understand disease pathogenesis.It will significantly accelerate the development of more specific and sensitive Tau tracers for achieving a differentiated diagnosis of tauopathies.
4.Conclusion
This review summarized the representative Tau-selective PET tracers, and categorized them into several chemical scaffolds.Most first-generation tracers shared structural similarities with Aβimaging tracers.Thus the modification was focused on enhancing Tau binding affinity and selectivity.
Numerous preclinical and clinical studies have shown that types of nitrogen-embedded heterocyclic scaffolds hold excellent properties for detecting Tau burdens.[18F]21, as the most representative tracer, has been approved for clinical diagnosis of AD in 2020.Subsequently, more second-generation tracers of novel scaffolds were developed to improve selectivity and avoid off-target binding to MAO.It is encouraging that some of them have successfully entered clinical studies, such as [18F]22, [18F]24, [18F]32, and [18F]35.At the same time, more human PET studies have found that these second-generation tracers have many other off-targets in the brain,which is also a considerable negative factor affecting imaging sensitivity.Hence, in-depth computational modeling studies need to be established.To our knowledge, the cryo-EM structures of Tau fibrils in various tauopathies have been successively elucidated,which will allow in silico simulation and determination of different binding sites on Tau fibrils.The computer-aided molecular design will significantly facilitate the discovery, characterization, and optimization of Tau radiotracers.More importantly, there is an urgent need to establish a suitable and consistent standard for Tau-PET tracers screening, which could better assist us in achieving the horizontal comparison among various tracers.
Although Tau tracers have demonstrated diagnostic applicability in AD, the new generation of tracers targeting specific Tau isomers remains to be developed.In addition, more horizontal comparative and longitudinal studies are needed to further evaluate the reported tracers, which could deepen our understanding of tauopathies.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This work was funded by the National Natural Science Foundation of China (Nos.U1967221, 22022601).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.024.
杂志排行

Chinese Chemical Letters的其它文章
- Professor Zhifang Chai: Scientific contributions and achievements
- Stable isotope labeling of nanomaterials for biosafety evaluation and drug development
- Emerging nanozymes for potentiating radiotherapy and radiation protection
- Recent progress of astatine-211 in endoradiotherapy: Great advances from fundamental properties to targeted radiopharmaceuticals
- 64Cu radiolabeled nanomaterials for positron emission tomography(PET) imaging
- Radiolabeled peptide probe for tumor imaging