欧盟关于食品动物用药理活性物质残留分析执行条例(EU)2021/808概述
2022-07-02王亦琳黄耀凌王鹤佳
孙 雷,王亦琳,叶 妮,尹 晖,张 骊,黄耀凌,王鹤佳
(中国兽医药品监察所,北京 100081)
作为欧洲农业食物链治理的关键要素,欧盟官方控制规则被全世界公认为最佳实践的典范。欧盟法规2002/657/EC[1]对药物残留分析方法的实施以及对活体动物和动物产品中药物残留分析结果的解释提出了要求,98/179/EC[2]规定了监测活体动物和动物产品中药物残留的官方采样详细规则。这两项决议都是在被废除的理事会指令96/23/EC[3]的基础上通过的,而且2002/657/EC不适用于在其他欧盟立法中制定了更具体规则的物质:食品中真菌毒素[4]、食品中二噁英和二噁英样多氯联苯(polychlorinated biphenyls,PCB)[5]以及食品中铅、镉、汞和苯并(a)芘[6]。鉴于新的科学发展以及法规的确定性和完整性,法规2002/657/EC和98/179/EC应废除,并被新的法规取代。2021年3月22日,欧盟官方公报发布了欧盟委员会执行条例(EU)2021/808《食品动物用药理活性物质残留分析方法的性能、结果解释和采样方法》,该条例替代2002/657/EC和98/179/EC,自欧盟官方公报发布后第20日即从2021年5月21日起生效。该法规的发布实施,对我国从事动物性食品等产品中药物残留分析有很强的借鉴意义。
1 主要内容
(EU)2021/808包含正文及两个附件,正文主要包括8个方面的内容:主题和范围、术语定义、分析方法的要求、实验室质量控制规定、结果解释、采样要求、废除法规和过渡措施、生效等。附件1列出了常用分析方法的性能标准和要求;附件2规定了动物性食品的官方采样程序和处理方法。
主题和范围中规定该条例适用于活体食品动物、动物身体部位和体液、排泄物、组织、动物源产品、动物副产品、饲料和饮水中药理活性物质残留的采样、实验室分析方法以及分析结果的解释。
术语定义中重点规定了49个术语定义:绝对回收率、准确度[7]、弃真误差(α误差)、被分析物、批准用物质[8]、去伪误差(β误差)、偏差、校正标准、有证标准物质(certified reference material,CRM)[9]、共色谱法(co-chromatography)、协作研究、确证方法、包含因子(k)、确证时的判断限(CCα)、筛查时的检测能力(CCβ)、添加样品、实验室间研究、内标(internal standard,IS)、关注浓度水平、最低校准浓度(lowest calibrated level,LCL)、基质、基质效应、基质匹配标准溶液、基质添加标准溶液、被测变量、测量不确定度、性能标准、精密度、定性方法、定量方法、回收率、回收率校正、标准物质[10]、相对基质效应、重复性、重现性[11]、耐用性、筛查方法、筛查目标浓度、选择性、单一实验室研究或内部验证、标准加入法、标准被分析物、物质、试验部分、正确度、单位、验证[12]、实验室间重现性或中等精度/内部重现性等。
实验室质量控制、分析结果解释以及采样方法等内容,世界各国都有各自相应的法规文件规定,这里不再一一表述。本文重点介绍(EU)2021/808中动物性食品等产品中药物残留分析常用的筛查方法和确证方法的性能要求。
2 筛查方法的性能要求
筛查方法包括生物、生化或理化筛查方法,包括用作筛查的定性、半定量或定量方法。
对于禁用或未批准用药物,筛查时的检测能力(CCβ)应尽可能低,在任何情况下,CCβ都应低于(EU)2019/1871规定的已建立行动参考点药物的行动参考点(reference point of action,RPA)。对于批准用药物,CCβ应低于MRL(maximum residue limit)或ML(maximum level)。只有能够以文件可追溯的方式证明经过了验证且假阴性率(β误差)小于或等于5%的分析方法才能用于筛查。如果出现可疑的阳性结果,则应通过确证方法进行确证。
用于筛查或确证的定量筛查方法应满足下面3.2项的准确度和精密度要求。
3 确证方法的性能要求
3.1 确证方法的一般要求 对于禁用或未批准用药物,CCα应尽可能低。(EU)2019/1871规定了RPA的禁用或未批准用药物,CCα应小于或等于相应的RPA。对于批准用药物,CCα应大于但尽可能接近MRL或ML。只有能够以文件可追溯的方式证明经过了验证且假阳性率(α误差)对于禁用或未批准用药物小于或等于1%,批准用药物小于或等于5%的分析方法,才能用于确证。
确证方法应提供分析物的化学结构信息。因此,仅基于色谱分析而不使用质谱检测的确证方法不适合用作禁用或未批准用药物的确证方法。对于批准用药物,在质谱法不适用时,可以使用其他方法,如HPLC-DAD法和HPLC-FLD法或它们的组合。
使用确证方法时,在提取步骤开始前,应添加适当的内标物。根据可用性,应使用特别适用于质谱检测的稳定同位素标记形式的分析物,或与分析物在结构上密切相关的类似化合物。当没有合适的内标物时,分析物的鉴别最好通过共色谱法进行确认。在这种情况下,只能获得一个峰,增强的峰高(或面积)相当于添加的分析物的量。如果不可行,则应使用基质匹配或基质添加标准。
3.2 确证方法的通用性能要求 准确度(accuracy)是指检测结果与公认的真实参考值之间的接近程度,通过正确度和精密度来确定。正确度(trueness)是指从一系列测试结果中获得的平均值与公认的参考值之间的一致程度。精密度(precision)是指在规定条件下获得的独立测试结果之间的一致程度,以测试结果的标准差或变异系数(coefficient of variation,CV)表示。
3.2.1 正确度(trueness) 对于有证标准物质的重复分析,实验确定的回收率校正平均质量分数与标准值的偏差应符合表1的最小正确度范围。当没有合适的CRM时,可以通过其他方式评估测量的正确度,例如使用具有实验室间研究指定值的物质或使用已知量的分析物添加到空白基质中来进行评估。

表1 定量方法的最小正确度Tab 1 Minimum trueness of quantitative methods
3.2.2 精密度(precision) 在实验室内重现性条件下,对标准或添加物质进行重复分析的变异系数(CV)不得超过Horwitz方程计算的水平。对于小于120 μg/kg的质量分数,应用 Horwitz 方程会产生不可接受的高值。因此,允许的最大变异系数不应大于表2中的值。在重复性条件下进行的分析其变异系数应小于或等于表2所列值的三分之二。

表2 可接受的变异系数Tab 2 Acceptable coefficient of variation
3.2.3 色谱分离要求 对于液相色谱法(liquid chromatography,LC)或气相色谱法(gas chromatography,GC),分析物的最小可接受保留时间应为色谱柱死体积保留时间的两倍。分析物在提取物中的保留时间应与校准标准、基质匹配标准或基质添加标准的保留时间一致,偏差为±0.1 min。对于保留时间小于2 min的快速色谱,保留时间的偏差小于5%。如果使用内标,分析物的保留时间与内标物的保留时间之比(即分析物的相对保留时间),应与校准标准、基质匹配标准或基质添加标准的相对保留时间一致,GC法的最大偏差为0.5%,LC法的最大偏差为1%。
3.3 质谱分析的特殊性能标准和其他要求
3.3.1 质谱检测 质谱检测应使用下列方式进行:(1)全扫描(full scan,FS);(2)选择离子监测(selected ion monitoring,SIM);(3)串联质谱(MSn)技术,如选择反应监测(selected reaction monitoring,SRM);(4)质谱(MS)或串联质谱(MSn)技术与适当电离模式的组合。低分辨率质谱(low-resolution mass spectrometry,LRMS,单位质量分辨率)和高分辨率质谱(high-resolution mass spectrometry,HRMS),包括如双聚焦、飞行时间(time of flight,TOF)和轨道阱质谱都是合适的。
在使用高分辨率质谱(HRMS)确证分析物时,所有诊断离子的质量偏差应小于5 ppm(或在m/z<200时低于1 mDa)。同时,应选择合适的有效分辨率,并且对于整个质量范围在10%峰谷处的分辨率通常应大于10000或半峰全宽(FWHM)处大于20000。
质谱检测时前体离子(precursor ion)应为准分子离子、分子离子的特征加合物、特征产物离子或其中一种同位素离子。非选择性的碎片或产物离子(如失水离子)应尽可能不选。选用的特征离子的信噪比(S/N)≥3。待确证分析物的离子相对丰度应与在相同条件下浓度相当的基质匹配标准、基质添加标准或标准溶液的离子相对丰度一致,偏差在±40%以内。
3.3.2 鉴别(identification) 应使用鉴别点系统选择适当的采集模式和评价标准。为了确证基质中已有MRL的药物,至少需要4个鉴别点(identification points,IP);对于禁用或未批准用药物,需要5个鉴别点(IP),其中一个鉴别点须来自色谱分离。
所有质谱法都应与一种色谱分离技术相结合,适合的分离技术包括液相色谱(LC)、气相色谱(GC)、毛细管电泳(capillary electrophoresis,CE)和超临界流体色谱(supercritical fluid chromatography,SFC)。如果分析物含有同分异构体化合物,保留时间的可接受性(即GC法为±0.5%,LC法和SFC法为±1%)是确证其存在的强制性要求。
表3所示为每种技术产生的鉴别点。为了获得确证分析所需的鉴别点,通过不同技术获得的鉴别点可以加和,最多可以组合三种单独的技术来满足最少鉴别点。不同的电离模式(例如电子电离和化学电离)被认为是不同的技术。表4给出了特定技术和技术组合得到鉴别点的示例。

表3 每种技术的鉴别点(IP)Tab 3 Identification points per technique
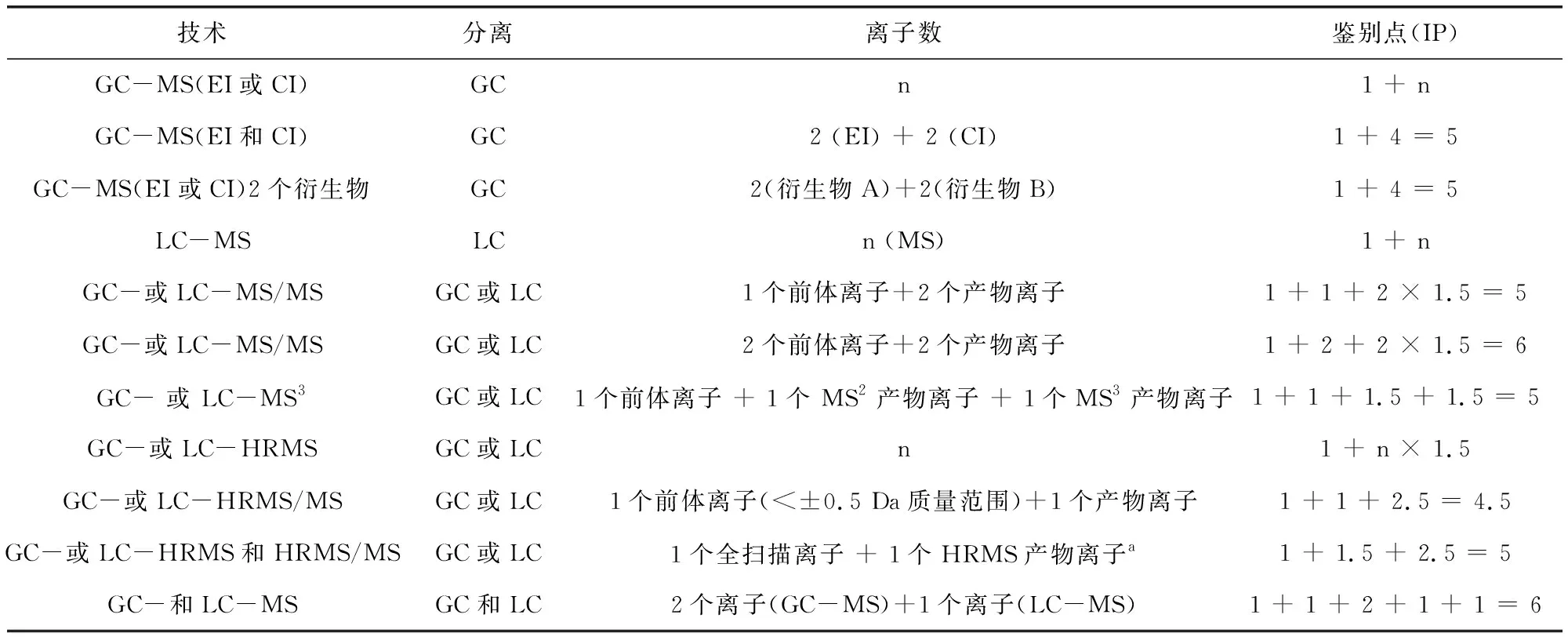
表4 特定技术和技术组合得到鉴别点的示例(n=整数)Tab 4 Examples of the number of identification points specific techniques and combinations of techniques (n=an integer)
3.4 质谱以外的检测技术特殊性能要求 仅对于批准用药物而言,下列技术可用作质谱法的替代方法,但须符合这些技术的相关要求:(1)全扫描二极管阵列检测分光光度法(diode array detection spectrophotometry,DAD)与HPLC一起使用;(2)荧光检测分光光度法(fluorescence detection spectrophotometry,FLD)与HPLC一起使用。具有UV/VIS检测器(单波长)的液相色谱本身不适合用作确证方法。
3.4.1 全扫描二极管阵列分光光度法的性能标准 除了满足色谱分离性能标准之外,分析物的紫外最大吸收应与基质中校准标准的波长在最大允许范围内保持一致,最大允许范围由检测系统的分辨率决定。对于DAD,最大波长范围通常在±2 nm以内。如果两个光谱比较中存在相对吸光度大于或等于10%的时候,那么在220 nm以上的分析物的光谱应与校准标准的光谱不应有明显差异。
当二者最大吸收波长相同,且两个光谱之间的差异不大于校准标准吸光度的10%,则认定被分析物满足标准。在使用计算机搜索和匹配谱库时,样品中的光谱数据与校准溶液的光谱数据比较必须超过临界匹配因子,该临界因子应在每个分析物的验证过程中根据满足上述标准的光谱来确定。验证过程中,还应检查由样品基质和检测器性能引起的光谱变化。
3.4.2 荧光检测分光光度法性能标准 除了满足色谱分离性能要求之外,结合色谱条件选择激发和发射波长时,应尽量减少空白样品提取液中干扰成分的影响。在激发波长和发射波长之间应该至少有50 nm的距离。色谱图中最近的最强峰应与指定的分析物峰至少相距一个分析物最大峰高10%处的全峰宽。此规定适用于具有天然荧光和在转化或衍生化后表现出荧光的分子的测定。
4 结束语
目前,国内兽药残留检测方法标准的制修订以及检验检测任务的实施等工作中分析方法的性能要求都以欧盟法规2002/657/EC为基础,其中色谱分离保留时间、离子鉴别点数要求、定性离子相对丰度等重要定性依据更是照搬欧盟这一法规。但欧盟法规2002/657/EC已于2021年废除,新发布的欧盟法规2021/808对色谱分离保留时间、离子鉴别点数要求、定性离子相对丰度等进行了修改,变得更加科学,更易操作,因此,对国内相关工作的开展具有很强的借鉴意义。
