Diagnosis and treatment of icteric hepatitis caused by erythropoietic protoporphyria: A case report☆
2022-06-30HanqingHuangLeiqinCaiXinhuaLiShuruChen
Hanqing Huang , Leiqin Cai , Xinhua Li , Shuru Chen
a Department of Infectious Diseases and Key Laboratory of Liver Disease of Guangdong Province, The Third Affiliated Hospital of Sun Yat-sen University,Guangzhou, Guangdong, China
b Department of Gastroenterology, People's Hospital of Longhua District of Shenzhen, Shenzhen, Guangdong, China
c Guang Dong Clinical Research Center for Metabolic Diseases, Department of Endocrinology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University,
Guangzhou, Guangdong, China
Keywords:Erythropoietic protoporphyria (EPP)Inherited disease Photosensitive dermatitis Jaundice Ferrochelatase (FECH)Liver injury Icteric hepatitis
ABSTRACT
1. Introduction
Erythropoietic protoporphyria (EPP, MIM 177000) manifests pseudodominant inheritance owing to ubiquitous loss-of-function polymorphisms in the intron and should be better recognized as recessive inheritance.1-4The EPP prevalence rate varies worldwide.As calculated,the EPP prevalence in Europe is 9.2 per million,25.4 per million in the United Kingdom, and 3.15 per million in Italy.5,6There are no epidemiological data related to EPP in China.
EPP is a hematoporphyrin metabolism disorder most commonly caused by pathogenic mutations in the ferrochelatase(FECH)gene,which encodes the terminal enzyme in heme biosynthesis.7For heme formation, which mainly occurs in the bone marrow and liver, mutations in FECH impair its activity and lead to the accumulation of protoporphyrin IX (PPIX) in the bone marrow,erythrocytes, and liver. It has been estimated that up to 20% of patients with EPP have liver injury, and approximately 2-5%develop serious liver damage or even liver failure.8Therefore,EPPassociated hepatopathy can be a prognostic factor, and regular follow-up of hepatic involvement is essential for EPP patients.
The clinical presentation of EPP varies. In infancy or early childhood,the skin manifestations as painful photosensitivity with itching, stinging, and burning sensations when exposed to strong sunlight.9In addition, the progression of photosensitive skin lesions depends on the intensity and duration of exposure to sunlight. If exposed to sunlight for a long time, erythema and edema will appear,and blisters or bullous lesions unique to skin porphyria will manifest in some cases.10Another clinical manifestation of acute porphyria, including EPP, is episodic acute neurovisceral attacks that can be life-threatening, with symptoms such as severe abdominal colic pain, nausea, vomiting, tachycardia, neurological manifestations, and others.11,12Nevertheless, severe liver damage has rarely been reported. Herein, we report a case of icteric hepatitis caused by EPP and review the relevant literature to improve our understanding of the clinical manifestations, pathogenesis,diagnosis and treatment of this disease.
2. Case report
The study was approved by the Ethics Committee of the Third Affiliated Hospital of Sun Yat-sen University.And written informed consent was obtained from the patient's parents in this case.A 15-year-old Chinese boy presented with photosensitive dermatitis,jaundice, abdominal pain during previous month when he visited our department again on April 20, 2021.
Skin stinging, itching, and burning in exposed areas are the initial symptoms experienced by the patient during approximately 10 minutes of sun exposure about one month ago.Then,the back of his hands presented with blister and getting bigger (Fig. 1). The patient also suffered from severe abdominal pain,persistent fever,cough, and jaundice, and his urine was dark yellow. He visited a local hospital for help. Comprehensive examination revealed elevated liver enzyme and serum bilirubin levels. Although the white blood cell counts were slightly higher than normal (Table 1,Phase I), no other signs related to infection were found, including screening for bacteria or viruses.
To make a definitive diagnosis, he was referred from local hospital to our hospital at first time on March 29,2021.A review of the patient's medical history showed that he had suffered from severe painful photosensitive dermatitis and jaundice for approximately 10 years. When exposed to strong sunlight for 30 min to 1 h, the sun-exposed skin starts to experience burning pain and itching,followed by swelling, redness, or blistering. Meanwhile, his urine appeared dark yellow or tea colored.It usually happens 2-3 times every year. Mostly, he would be relieved after rest for about three days without joint malformation, joint pain, or pigmentation. His parents were healthy and without photosensitive dermatitis or jaundice. However, his younger brother also developed photosensitive dermatitis.
Although he had taken antibiotics, liver protection drugs, and other drugs for almost one month,his condition did not ameliorate.During hospitalization, skin damage gradually become healed,leaving dark brown pigmentation(Fig.1).However,abdominal colic pain worsened described as high intensity,frequently occurring at midnight without radiation pain, aggravated after defecation, and relieved after spasmolysis and acetanilide.Moreover,his urine was still tea-colored more than ever, and experienced paroxysmal tachycardia sometimes. As the patient's condition worsened,further examination was performed.Serum biochemistry revealed that the liver enzyme and serum bilirubin levels were significantly higher than before (Table 1, Phase II), but the following results revealed no abnormalities, including viral and autoimmune hepatitis-related antibodies,rheumatic immune related antibodies,blood lead, urine lead, and blood zinc protoporphyrin (ZPP)(Supplementary Table 1). Computed tomography suggested slight dilatation of the intrahepatic bile duct,enlarged hilar lymph nodes,and slightly enlarged liver and spleen. After multidisciplinary consultation,intestinal perforation,intestinal obstruction,bile duct obstruction, and other diseases were excluded. To summarize, the typical clinical manifestations, including painful photosensitive dermatitis, neurovisceral symptoms, non-organic abdominal colic pain,jaundice,and genetic testing revealed two mutation sites in the FECH gene(Exon8 c.892C >T(p.R298X),heterozygous;Exon4 c.315-48T >C (splicing), homozygous), which confirmed the diagnosis of EPP.13,14
On April 20,2021,he received a glucose load(400 g,once daily(qd)), corresponding to ursodeoxycholic acid (500 mg, twice daily(bid)) and cholestyramine (5 g, bid) regularly. As a result, the patient's symptoms disappeared, with no recurrent abdominal pain or paroxysmal tachycardia,and jaundice gradually decreased. Two month later,his skin of face,neck,and both hands become normal without pigmentation (Fig. 1). Concomitantly, liver enzymes and bilirubin levels all returned to normal (Table 1, Phase III) (Fig. 2).
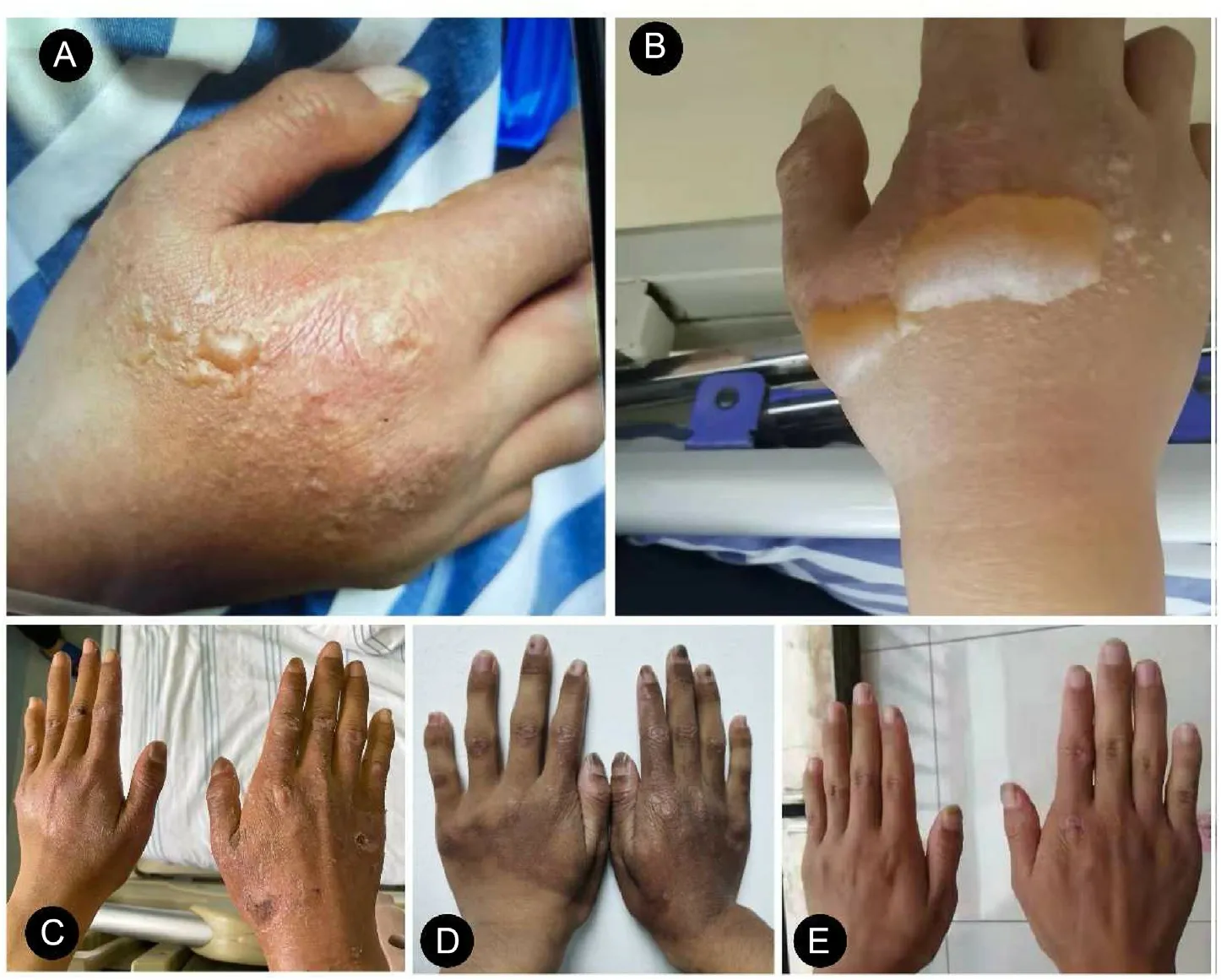
Fig.1. Follow-up the development of the patient's photosensitive dermatitis.(A)Initial stage(March 15,2021).(B)Acute stage(March 17,2021).(C)Healed stage(April 2,2021).(D) Recovery stage (April 27, 2021). (E) Normal (June 17, 2021).

Table 1 Biochemical tests of the patient.
3. Discussion
EPP is a rare autosomal recessive disease that typically manifests as painful photosensitive dermatitis, jaundice, and neurovisceral.EPP diagnosis should be based on a comprehensive analysis of classical clinical presentations,positive laboratory findings such as increased levels of porphobilinogen (pp) in plasma, feces and erythrocytes, and the detection of a plasma fluorescence peak at about 630-634 nm.10,15Furthermore, the diagnosis of EPP is confirmed by mutation of the FECH gene, as revealed by genetic sequencing.9
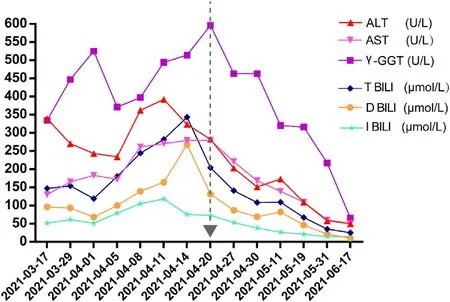
Fig.2. Long-term follow-up of the patient's biochemical parameters.Confirmatory diagnosis on April 20,2021,the start of treatment with glucose load(400 g,once daily(qd)), corresponding to ursodeoxycholic acid (500 mg, twice daily (bid)) and cholestyramine (5 g, bid) regularly until June 17, 2021. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; DBIL, direct bilirubin; γ-GGT, gammaglutamyl transpeptidase; IBIL, indirect bilirubin; TBIL, total bilirubin.
With the popularization of gene sequencing,most patients with typical clinical manifestations of EPP have mutations in the FECH allele and low expression of c.315-48C (also called IVS3-48C) mutations.16In this case, the patient has two mutations in the FECH gene, c.892C >T (p.R298X, heterozygous) and c.315-48T >C (homozygous). Although in some cases, the homozygous c.315-48C variants showed no biochemical or clinical symptoms of EPP, the variant c.892C >T (P.R298X, in Exon 8) in the heterozygous state has been detected in recessive inheritance EPP patients in a previous study.13Since his parents' sample were not obtained, the genetic variant sources were not verified. Therefore, we enquired the patient's family history. His parents were healthy and denied any symptoms related to EPP,whereas his younger brother had the same manifestations of EPP. Notably, photosensitive dermatitis caused by EPP should be distinguished from phototoxic drug reactions,chickenpox,solar urticaria,contact dermatitis,and vascular edema.10Therefore,a prompt skin biopsy should be performed for cases with atypical presentations.Neurovisceral symptoms such as abdominal pain should be distinguished from other metabolic causes, such as lead poisoning or the rare 5-aminolaevulinic acid dehydratase porphyria, by measuring 5-aminolaevulinic.17The liver function abnormalities include drug-induced liver injury,viral hepatitis,autoimmune liver disease,primary sclerosing cholangitis,and progressive familial intrahepatic cholestasis.A liver biopsy may serve as a distinguishing diagnosis of EPP.The features of EPP reveal a bright red birefringent centrally located Maltese crossed accumulated within hepatocytes and Kupffer cells under polarized light.18
For patients with EPP,photosensitivity may impact their quality of life. Acute porphyria, including EPP, can be manipulated by avoiding precipitating factors, such as direct sun exposure, porphyrinogenic drugs (chloroquine, sulfonamide antibiotics, rifampicin),barbiturics,estrogens and progestins,alcohol,and fasting.19Additionally, cutaneous porphyria, a photosensitivity disorder,relied on sunlight avoidance. The concomitant use of protective clothing or physical sunscreens containing zinc or titanium oxide can block ultraviolet and visible light to reduce the occurrence of photosensitivity.7The afamelanotide (Scenesse®) is a synthetic alpha-melanocyte-stimulating hormone analogue and first-in-class melanocortin-1 receptor agonist, which can be used to prevent phototoxicity and reduce pain caused by direct sunlight in adult patients with EPP.20Oral beta-carotene(120-180 mg per day)has been used to improve light tolerance to sunlight, but the available data are insufficient.21In addition, vitamin D and calcium supplementation is recommended for patients with EPP because sunlight exposure is avoided.22
Hepatobiliary disease associated with EPP may contribute to an unoptimistic prognosis. Due to the deficiency of FECH activity in EPP patients, PP, which is insoluble in water, increases and then accumulates in the liver. Excessive PP induces oxidative stress injury of hepatocytes. Therefore, reducing PP accumulation in the liver plays a crucial role in the treatment of EPP-related hepatobiliary diseases.10,23Cholestyramine not only helps to interrupt enterohepatic circulation of PP but also may bind PP to promote faecal excretion.24Ursodeoxycholic acid is administered to increase the secretion of PP into bile.18,25However, a previous study has suggested that ursodeoxycholic acid may only be effective in the primitive phase of liver disease in EPP. Liver transplantation is the common treatment of choice for patients with end-stage liver disease.25However,owing to the inability to reduce the production of PPs,recurrence will occur sooner or later,and the ideal treatment might be consecutive liver and bone marrow transplantation, as reported so far.26Remarkably, special optical filters should be utilized to minimize EPP patients’exposure to visible light during liver transplantation.27Plasma exchange can be performed as a bridge to liver transplantation to remove PP.28
Acute porphyria,including EPP,may present with a sudden lifethreatening crisis manifesting as severe abdominal pain, neuropsychiatric symptoms, and other symptoms.29No matter which type of porphyria, hematin infusions, glucose or carbohydrate loading are methods for both the treatment of acute attacks and for the prevention of recurrent attacks.19,21,30,31The therapeutic effect of glucose in patients has been well documented.Previous studies have reported that glucose loading can significantly reduce the production and excretion of PP and amelioration of symptoms.21,32This case illustrated that after two-month treatment with cholestyramine, ursodeoxycholic acid, and glucose loading, the liver function significantly recovered, and the symptoms were gone without skin pigmentation, joint deformity, and facial changes;besides, severe abdominal pain never occurred again.
EPP is a lifelong disorder whose prognosis depends on the evolution of hepatic disease,therefore,regular follow-up of hepatic involvement is essential.10It is recommended that patients with EPP undergo regular re-examination,including blood biochemistry(liver function), tumor markers, and abdominal ultrasound. In addition,genetic sequencing should be performed in patients with typical symptoms of EPP, if economic conditions are permitted.Furthermore, pathogenic gene mutations are identified in an affected family proband, and screening of families to identify presymptomatic carriers is also recommended to decrease the risk of overt acute porphyria. Moreover, genetic counselling and prenatal diagnosis are recommended for high-risk pregnant women with a family history of EPP.
Authors’ contributions
S. Chen and X. Li designed the case report, obtained research funding, and revised the manuscript. H. Huang collected the patient's clinical data,analyzed the data,and drafted the manuscript.L. Cai analyzed and interpreted the data and revised the manuscript. All authors read and approved the manuscript for publication.
Declaration of competing interest
The authors declare that they have no conflicts of interest.
Acknowledgements
This work was supported by the Guangdong Key Field R&D Plan of China (2019B020228001) and the 5010 Project of Clinical Research in Sun Yat-sen University, China (No. 2018024).
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.livres.2022.05.003.
杂志排行
Liver Research的其它文章
- Expert consensus on the role of hematological markers in the early clinical screening of hepatocellular carcinoma☆,☆☆,☆☆☆
- Camrelizumab (SHR-1210) treatment for recurrent hepatocellular carcinoma after liver transplant: A report of two cases☆
- Quantification of liver fat deposition in obese and diabetic patients: A pilot study on the correlation with myocardium and periapical fat content☆
- Protective effects of Longhu Rendan on chronic liver injury and fibrosis in mice☆
- Effects of intestine-specific deletion of fibroblast growth factor 15 on alcoholic liver disease development in mice☆
- Non-alcoholic fatty liver disease development: A multifactorial pathogenic phenomena☆