单原子催化剂在电催化还原CO2领域的应用
2022-06-29金湘元张礼兵孙晓甫韩布兴
金湘元,张礼兵,孙晓甫,韩布兴
(中国科学院化学研究所,胶体、界面与化学热力学实验室,北京 100190)
能源问题是当今人类社会面临的重大问题之一[1~3].目前,世界上的能源供给主要由化石燃料的利用来实现,而化石燃料是一类不可再生能源,不断消耗的同时也带来了全球变暖,海平面上升等诸多威胁人类安全的问题[4].CO2转化利用是对人工碳循环进行闭环,从而实现碳中和的有效方式[5].在诸多转化方法中,电催化CO2可以在较为温和条件下,将其它可再生资源产生的间歇性能量[6](如风能、潮汐能和太阳能等)以化学能的方式储存起来,同时获得高附加值的化学品,符合可持续发展的需要.
单原子催化剂是以孤立原子为催化活性中心,并通过配位锚定在载体上的负载型单分散催化剂,与体相催化剂不同的是,其不存在金属原子聚集体.单原子催化剂由于独特的电子结构和最大化的原子利用率被广泛应用在各个领域.自2011年首次提出单原子催化的概念[7]以来,国际学术界对单原子材料进行了广泛深入的研究,包括其在能源转化[8]、疾病治疗[9]和环境保护[10]等领域的应用.锚定在多孔碳上的金属-氮键活性位点通常是单原子催化剂最主要的存在形式.这种金属与氮之间的化学键类似于溶液中配位化合物中配体与金属离子之间的配位键[11].实现这种结构主要是利用含氮有机配体和金属前驱体的相互作用,然后在高温碳化过程中金属-氮键被保留下来并嵌入碳基质中.利用金属有机框架材料(Metal-Organic Frameworks,MOFs)的规则孔道来封装和分散金属离子是比较常见的方法,通过高温热解(800~1000 ℃),实现有机配体的碳化,同时金属离子也在碳化过程中被还原.封装的金属离子与氮原子之间的键一般不会断裂,获得M-Nx的结构(其中M代表金属原子),实现了单原子的分散,这种限域的方法通常在高温也能防止团簇的产生[12].
CO2分子中存在两个的离域π键,C=O 键能为806 kJ/mol[13].碳元素的化合价为+4 的最高价态,表明还原CO2是一个需要较高活化能的缓慢动力学过程[14].同时,由于CO2在水中的溶解度较低(约34 mmol/L),传质过程也将显著影响CO2催化还原的进程.在催化剂表面,CO2活化大概涉及4种反应方式[15]:
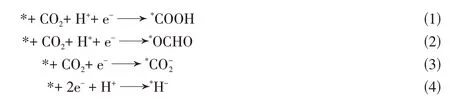
*COOH进一步还原生成*CO,而*OCHO进一步还原生成HCOOH.式(3)为CO2的非质子耦合活化过程,式(4)中表示的吸附H—是对CO2还原过程中的中间体进行亲核进攻的,是生成多电子产物的必需中间体.后续在催化剂表面还会通过吸附的C1中间体的C-C偶联来实现C2+产物的生成,这样CO2还原就有可能生成多种不同的产物.因此,电催化CO2存在低法拉第效率(Faradic Efficiency,FE)、低电流密度(Current density,j)以及高起始电位(Onset potential)的问题.
目前,CO2电催化剂主要包括金属单质[16]、金属化合物(如氧化物[17]、氮化物[18]、硫化物[19]等)、无机有机杂化材料(如COF[20],MOF[21]等配合物)及单原子催化剂[22].然而,这些催化剂存在只利用表面原子而导致的活性位点较少,选择性较低,催化活性位点不明确等现象,这些问题的存在表明亟需设计新型催化剂.近期的单原子催化剂作为复相催化剂,成为了沟通复相催化剂和均相催化剂之间的桥梁,具有易于回收、活性位点多且明确、电子结构可调的特性,可以设计具有独特电子结构的活性位点,以对目标产物对应的中间体产生特异性吸附,从而实现高活性和高选择性CO2还原.本文综合评述了从过渡金属到主族金属等非贵金属单原子,以及通过杂原子配位、双/单原子位点、金属-载体相互作用、空间限域和分子桥联等策略,来调控单原子催化剂的微环境,从而实现高活性和选择性电催化CO2还原,且具有较好的稳定性.最后,还对单原子催化剂在CO2电催化还原领域的发展进行了展望.
1 金属单原子催化剂
1.1 过渡金属单原子催化剂
基于过渡金属制备的催化剂在电催化CO2还原领域被研究得最多且最深入.过渡金属由于存在未被电子占据的d轨道,其价电子的电子结构在与CO2还原中间体发生特异性吸附时发生改变.电催化CO2还原是一个多步电子/质子耦合反应过程,催化剂的本征活性由催化剂表面活性位点的电子行为决定,在此情况下,过渡金属价电子结构的变化有利于提高电化学CO2的还原效率和选择性.
1.1.1 Fe-N-C 由于铁(Fe)是地球上丰度第二高的金属,因此,发展Fe基单原子催化剂实现CO2电还原较为可取.也正因为其较大的丰度,以最常见的Fe-Nx结构,来研究CO2在此类材料上发生还原反应的机理就变得较为方便.但是单原子催化剂的制备过程中存在单原子与纳米颗粒共存的常见问题,对催化性能产生不可忽视的影响.通过调整Fe-N4位点与Fe 纳米颗粒的比例,可以调整CO/H2的产物比例.其中不含有Fe纳米颗粒的,Fe质量负载量为1.5%的Fe-N-C催化剂(Fe0.5d)性能最好,说明Fe-N4是催化CO2到CO的活性位点[23].也有研究利用与Fe位点亲和性比较好的反应物(如SCN-),在不影响N-C位点的情况[24]下,通过与Fe-N4特异性结合来毒化活性中心,证明了与吡啶氮结合的Fe位点是CO2还原的真实活性位点[25].目前,Fe-Nx结构通常都是以MOF作为碳框架(如ZIF-8),然后进一步吸附铁离子,利用高温下Zn挥发产生空位捕获Fe而得到Fe-Nx的结构.Gu等[26]通过在900 ℃下热解Fe掺杂的ZIF-8,得到了以Fe-N结构固定在碳骨架上的原子分散的Fe3+,并进一步通过催化活性位点的辨别,说明了其氧化态对于催化活性的影响.其明显比700 ℃下得到原子分散的Fe2+表现出更快的反应动力学过程.前者塔菲尔斜率为64~71 mV/decade,后者为117 mV/decade.其起始电位为-0.20 V.在340 mV的过电位下,达到了94 mA/cm2的电流密度.在-0.47 V(vs.RHE)(Reversible Hydrogen Electrode,可逆氢电极)下,达到了FECO>90%.Fe3+位点是催化活性的关键,还可以从催化剂在-0.5 V下由于Fe3+还原为Fe2+从而活性下降而得到证明.另外,Li等[27]报道了原位生成的Fe(I)位点是CO2电还原活性位点.Fe(I)具有单电子的dz2轨道和[COOH]中单占据的π*轨道之间强烈的相互作用,是优异CO2RR活性表现的关键.对于生成需要多于2e-的还原产物,CO在Fe-N4位点上较大的脱附自由能反而可能起了积极的作用.同时,周围位点对*H的适当吸附正可以与结合在Fe位点上的CO结合,促进了需要多于2e-还原产物的形成[28,29].
有研究者从另外一个角度阐释了催化活性位点对催化活性的影响.除了阐明了单原子Fe 比单原子Co 有更高的电流密度和FECO,他们从计算角度阐释了连接两个相邻扶手椅型的石墨烯层的Fe-N2+2-C8位点,比嵌入石墨烯层中Fe-N4-C10构型具有更高的催化活性.*CO 的吸附发生在Fe 原子上,同时*COOH 的解离发生在Fe-N2+2-C8位点.N 原子周围具有悬挂键的碳原子是对*OH 进行吸附的活性位点.有利于*COOH 转化为*CO.这些因素导致了较高的法拉第效率[在−0.6 V(vs.RHE)下FECO>90%],−0.9 V 下可以达到10 mA/cm2的电流密度[30].对于Fe-N2+2位点而言,局部的电子效应平衡了*COOH和*CO的结合能,也可能是其具有较高催化活性的原因[31].
目前,较多的研究集中在阐明以Fe-Nx位点为催化的活性中心上,周围的环境对催化环境的影响经常被忽略.M-N-C和相应的金属-卟啉配合物的催化性能趋势不匹配,表明Fe-N4周围的碳骨架可能对催化活性有影响[32].其M-N-C的合成方法如图1(A)所示.Zhu等[33]发现,Fe-N4附近石墨层中的N位点通过降低*COOH 形成的能垒来加速CO2至CO 的转换.也有其它研究证明了周围环境中额外的N掺杂对于CO2还原的促进作用.通过在惰性气氛中掺入H2热解,以实现减少对质子亲和强的位点(非配位的吡啶氮和吡咯氮位点),从而抑制析氢反应,使得在Fe-N4/石墨型氮原子界面实现-0.3~-0.8 V(vs.RHE)下CO法拉第效率均在90%以上.这样就解决了以前研究中高法拉第效率区域对应的较窄电位窗口(−0.4~−0.5 V)中不包含*CHO 中间体形成的问题[34].值得一提的是,有研究指出,相比于Fe-N4位点,石墨烯碳平面上的缺陷才是催化活性位点,而Fe-N4位点与缺陷结合降低了CO2RR 的能垒[35].近期,也有研究人员关注于宏观条件(催化剂工作温度).Lin 和Li 等[36]报道Fe-N-C 和Ni-N-C 催化剂在303~343 K下展现出不同的性能表现,归结于关键中间体与Fe-N-C和Ni-N-C的结合强度不同.
目前,对于Fe-N-C上进行CO2还原的机理研究还存在争议,仍有待进一步的研究.虽然Fe-N-C显现出对CO较好的产物选择性和产率,但对于Fe-N-C如何更高产率地生成多电子转移产物,还有待进一步探索.
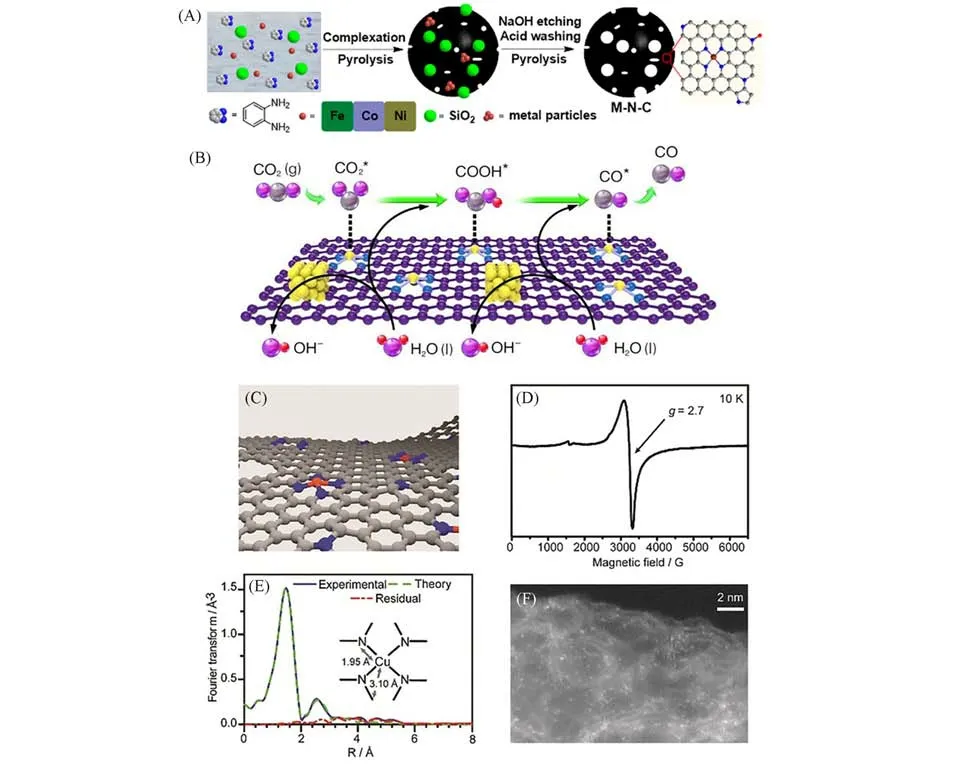
Fig.1 Protocol for the synthesis of M⁃N⁃C electrocatalysts(M=Fe,Co,Ni)(A)[32],a proposed reaction mechanism for CO production via ECR of Ni@NiN4CM(Ni yellow,N blue,O pink,C gray,H red)(B)[50],schematic representation(C),EPR spectrum record at 10K of Cu0.5NC diluted in an amorphous silica matrix(D),Cu K⁃edge EXAFS analysis in the Fourier⁃transformed space(E)and HAADF⁃STEM image of Cu0.5NC(F)[54]
1.1.2 Co-N-C 有早期研究显示,Co-N-C 在−0.6 V(vs.RHE)下,FECO≈45%;在−0.5 V(vs.RHE)下,FECO≈30%,均弱于相同测试条件下Fe-N-C 和Ni-N-C 的表现[37].然而,近期报道的Co 单原子催化剂的CO 选择性可达到90%以上[38,39],且Co 单原子活性比Fe 单原子更高.通过去除未与Co 单原子配位的N 原子,孤立的CoN4位点*COOH 形成能垒抑制了析氢反应的发生[38].因此,在H 型电解池中,0.49 V的过电位下,以-24.9 mA/cm2的电流密度达到了99.4%的CO法拉第效率.流动池中,在较宽的电流密度窗口范围(50~600 mA/cm2)内,FECO>90%.一种具有CoN4-CB 位点的单原子催化剂,其使用的一种Co盐与1,10-邻菲咯啉反应形成配合物,然后与碳黑混合均匀后共热解,最后酸浸得到了CoN4-CB[39].在-0.76 V下,其转换频率(Turnover Frequency,TOF)为27173 h-1,CO分电流密度为-33.6 mA/cm2.总的电流密度为98.3 mA/cm2下,FECO平均为92.1%.远远超过了同等测试条件下Ni-N4的TOF和电流密度(j).这种Co 单原子催化剂表现出反常的高CO 选择性,在-0.3~-0.75 V 的范围内,FECO(平均约为90%)超过了NiN4-CB.理论计算进一步阐明,由于Co-N 位点降低了*COOH 的形成能垒,并且*CO 的脱附能垒较小,导致了其催化性能较高.
1.1.3 Ni-N-C 因较高的CO产物选择性,Ni单原子催化剂近期有较多报道.首次报道用于电催化CO2还原的Ni 单原子催化剂,是通过MOF 的空洞中Ni2+与锌节点的离子交换后,高温热解MOF 而得到.TOFCO和FECO可分别达到5273 h-1和71.9%.其电流密度在0.89 V的过电位下达到了10.48 mA/cm2[40],通过热迁移策略,将在氮掺杂碳上Ni纳米颗粒在高温下原子化,氮掺杂碳中具有强配位作用的位点捕获Ni 原子,实现Ni 单原子催化剂的构筑[41].Ni 纳米颗粒原子化后所留下的孔洞可以促进溶解在电解液中的CO2与Ni 单原子位点接触.这在一定程度上解释了其具有突出性能的原因.其在-1.2 V(vs.RHE)的电位下,在H型电解池中达到了约30 mA/cm2.在−0.6~−1.0 V的电位窗口范围内,FECO≥90%,在-1.1 V(vs.RHE)的电位下,TOFCO=48842 h-1.热解含有碳涂层的Ni掺杂g-C3N4材料也实现了CO2到CO的转换.在-0.5~-0.9 V的电位下,Ni-N4-C的FECO超过90%,在−0.81 V下达到最高为99%.此电位下的电流密度为28.6 mA/cm2.X 射线吸收谱(X-ray Absorption Structure,XAS)表征证明了其Ni-N4位点的存在[42].另有研究者通过一种通用的种子策略,合成了一种固定在氧化石墨烯上的Ni 单原子(SA-GO).在较低的过电位−0.63 V 的条件下,TOF 为325.9 h-1,FECO约为96.5%[43].通过拟合X 射线吸收近边精细谱(X-ray Absorption Near-Edge Structure,XANES),证明了Ni-N4位点的存在.同时研究发现,改变碳基底的结构以构建介孔隧道,可以增强反应传质过程,来进一步提高Ni-N4的催化活性[44].其中,以热不稳定的5-氨基四氮唑为自牺牲模板,2-甲基咪唑为碳源和氮源,构建的负载在中空NC 骨架上的Ni-N4位点单原子催化剂(Ni-AHP),实现了在-0.7~-1.0 V 的电位窗口内FECO接近100%[45].
催化剂精细结构表征可以进一步探究Ni-N4活性位点在CO2RR中的作用.Yang等[13]合成了一种锚定在石墨烯上的Ni(i)-N4活性位点的单原子催化剂,并通过XAS,X 射线衍射谱(X-ray diffraction,XRD)和X射线光电子能谱(X-ray Photoelectron Spectroscopy,XPS)系统地证明了Ni(i)-N4的配位结构.在0.61 V的低过电位下,催化剂表现出CO2电催化还原的高本征活性.此电位下可以达到350 A/gcatalyst的质量活度,转换频率高达14800 h-1,并同时表现出较高的FECO(97%).在22 mA/cm2的电流密度下,持续电解100 h,催化剂仍保持着98%的法拉第效率,具有较高的稳定性.如此高的催化活性归结于Ni(i)位点独特的3d9的电子构型.3dx2−y2中未配对的电子发生了离域,同时发生从Ni(i)位点到CO2中C的2p轨道的电子转移,使得CO2转变为COδ−2,这是CO2电还原的第一步.值得提出的是,d9的电子组态可能是Ni(i)-N4的构型为扭曲平面正方形的原因,而石墨烯框架阻止了Ni(i)-N4位点的进一步扭曲,确保了CO 的快速脱附[11].也有研究表明,Ni(i)位点是CO2活化的催化活性位点.负载在碳纳米管上的Ni-TAPc中的Ni2+在电化学原位还原中得到的Ni+位点,对CO2活化有较高的活性,并且是真正的催化活性位点,这已经被原位(Operando)光谱和电化学动力学实验证明,Ni+与CO2键合并使之发生进一步活化.在0.6 V 的过电位下,达到了99%的CO 法拉第效率,电流密度j达到32.3 mA/cm2.TOF 达到100179 h-1,在目前的研究中处于较为领先的水平[46].
另有,研究也报道了几种Ni-N4位点具有促进CO2RR的作用.一种含有Ni-N4位点的Ni1-N-C模型催化剂,比相同合成方法得到的Fe1-N-C,Co1-N-C和Cu1-N-C具有更高的CO产物选择性[47].此外,还有一种新颖的离子吸附策略,避开了传统方法上需要的严苛条件(如高温和酸浸).热解g-C3N4和葡萄糖的混合物获得超薄碳纳米片并作为载体,通过在Ni(NO3)2水溶液中进行浸渍吸附,进一步在Ar气氛保护下,于300 ℃低温热处理来实现稳定吸附金属位点.所获得的催化剂在-0.68 V 下达到了大于90%的FECO.Yang 等[48]采用了电纺丝法制备了锚定在石墨烯上的Ni-N4催化剂.所获得的交联纳米纤维具有分层的孔隙结构,有助于提高Ni-N4活性位点密度,以促进CO2还原效率.进一步利用流动池测试得到了308.4 mA/cm2的CO电流密度.且在电解120 h后,其仍能保持88%的CO法拉第效率.此外,通过快速热解得到的含有非典型Ni-N4位点的氮化碳基催化剂(Ni-SAC@NCs)是另一种制备单原子的策略[49].该方法通过在管式炉上安装往复装置(从加热区到冷却区)来达到快速热解,过程中产生的内部高压阻止了单原子的团聚.值得一提的是,碳酸钠被用来促进多孔碳骨架的形成.在热解水洗前,其起到碳化过程石墨烯的模板和维持骨架的功能,水洗后还能实现材料的多孔性.因此,在流动池中催化剂的CO电流密度达到了187.7 mA/cm2.
近期,对于金属纳米粒子与金属单原子协同促进电催化的文献已偶有报道,表明近期学术界对于其研究兴趣的增长[23,50~52].Ni纳米颗粒加速了吸附H的形成,并将其传输给临近的Ni-N4位点,促进了CO2还原过程中中间体的质子化过程,因此促进了整体CO2电还原过程.后续研究也发现纳米颗粒能够促进CO2还原过程中的质子化过程[51].Cu-S1N3/Cux与Cu-S1N3相比,决速步骤CO2到*COOH 的反应能垒得到了降低.这归结于Cu-S1N3/Cux与Cu-S1N3对比,前者促进了*H2O到*H的转变,从而促进了CO2质子化到*COOH 的过程.协同的催化剂在较宽的电流密度下(约-0.6~-0.9 V 的电位下,实现了FECO≥50%),并达到了较高的电流密度[50].可能的协同机制如图1(B)所示.Ni-N4附近的ZrO2纳米颗粒(ZrO2@Ni-NC)加快了*CO−2的质子化到*COOH 的这一决速步骤.同时也促进了*COOH 质子化脱水生成*CO的过程.其CO2还原起始电位仅为−0.3 V,并在−0.7~−1.4 V的电位窗口范围内实现了大于90%的FECO,最高可达(98.6±1.3)%[52].
1.1.4 Cu 在电催化CO2RR领域,Cu基电催化剂是其中最具有前景的材料之一.Cu基催化剂具有可将CO2还原到多碳产物的能力.但不同于尺寸较大的体相Cu 基催化剂,Cu 单原子催化剂主要生成C1产物[51].这是由于不同的铜单原子之间距离较远,不利于形成C-C偶联产物.同时,理论计算表明,铜单原子的稳定性较差,在-0.7 V(vs.RHE)的条件下,铜单原子会发生迁移,并团聚为金属铜纳米颗粒[53].这也限制了其进一步应用.
然而,Karapinar 等[54]却报道了一种Cu-N4位点,可以实现CO2RR 生成乙醇.这种材料是通过ZIF-8、氯化铜和1,10-邻二氮菲,在固相中经过低能量的球磨实现混合,后续在1050 ℃和Ar气气氛下对前驱体进行碳化得到Cu0.5NC,其结合和形貌表征如图1(C)~(F)所示.如图可知,Cu以单原子分散的形式存在,并且配位结构为Cu-N4.在-1.2 V(vs.RHE)以及0.1 mol/L CsHCO3的条件下,实现了55%的乙醇法拉第效率.此催化剂也可以实现一氧化碳还原(CORR)生成C2产物(乙醇和乙烯).且C2总法拉第效率达到80%.有趣的是,研究发现在电解条件下,孤立的Cu单原子会瞬时转换为Cu金属纳米颗粒.与之前理论计算研究预测Cu单原子团聚成Cu纳米颗粒后电催化性能会下降的结论相悖[53].电解后的材料又恢复为Cu 单原子,Operando XAS 表征也进一步解释了这一现象.研究认为这种在电解过程中形成的Cu 纳米颗粒中间态,可能是真正的催化活性的来源[54].Xu 等[55]也发现电催化条件下铜单原子会形成Cu簇(含有3~4个铜原子),而铜簇是真正的催化活性位点.在-0.7 V(vs.RHE)下达到了91%的乙醇法拉第效率.这也是迄今为止由单原子Cu经过CO2RR得到乙醇产物的最高水平.另有研究人员通过追溯他们以前的相关研究,创造性地使用熔融的金属Li来溶解少量金属铜,在溶液中实现Cu的单原子分散,然后经过淬灭,得到了Cu-Li的双组分电催化剂.这种Cu-Li被放置在常规潮湿环境中会形成Cu-LiOH,后续与碳黑混合后用去离子水浸出LiOH,就可以得到均一分散的Cu 单原子催化剂.高角环形暗场-扫描透射电子显微镜(High-Angle Annular Dark Field-Scanning Transmission Electron Spectroscopy,HAADF-STEM)表征进一步证明了Cu的单原子分散[55].但由于催化剂本身的限制,所获得的产物(乙醇,丙酮)电流密度较小,大约仅为−1.8 mA/cm2.另有研究通过将Cu(OAc)2封装在ZIF-8的孔道中,后续通过热解得到了负载于氮掺杂多孔碳上的铜单原子催化剂,实现了CO2电催化还原生成C3产物丙酮(CH3COCH3,主要产物),法拉第效率达到了36.7%.CH3COCH3的产率为336 μg/h.DFT计算显示,与4 个吡咯型氮原子配位的Cu 单原子是最主要的活性位点,这种结构降低了CO2活化和C-C偶联的能垒,并可以稳定CH3COCH3产生过程中的中间体[56].目前,CO2还原生成CO可以达到较高的产率和选择性,但是多碳产物的选择性仍较低,研究通过在MXene(Ti3C2Tx)上负载Cu单原子实现了CO到多碳产物的转换,在-0.7 V(vs.RHE)下,多碳产物的选择性达到了98%,其中乙烯的法拉第效率为71%.可归结于Cu-O3位点发生CO的C-C偶联并生成*CO-CHO途径,此决速步骤的中间体形成能垒较低[57].这些铜基单原子催化剂的研究进一步展现了单原子材料应用于CO2RR所具有的独特优势.
1.1.5 Zn-N-C Zn-N-C单原子催化剂中Zn的含量通常较少,这是因为Zn具有较低的沸点(907 ℃),在有机配体碳化的同时也会导致Zn原子的挥发.Han等[58]通过二水合醋酸锌(锌源)、葡萄糖(碳源)和盐酸羟胺(氮源)在低于锌沸点的温度下(800 ℃)共热解,实现了Zn-N-C单原子催化剂的构建.不同于其它过渡金属单原子催化剂,该催化剂CO2RR的产物为CH4,并且CH4的法拉第效率达到了85%,部分电流密度为−31.8 mA/cm2,稳定性测试显示,电解35 h后电流密度和法拉第效率均未明显衰减.理论计算进一步表明,Zn-N-C位点上CO的生成能垒较高,而*OCH中间体的自由能较*CHO更低,更易质子化形成CH4.也有研究表明,共存的Zn-N4和Zn-N3位点的Znδ+-NC(0<δ<2)可以在仅310 mV 过电位下FECO达到近100%,并在-0.5~-1.0 V的电位窗口内保持高选择性.进一步在流动池中电解测试发现,催化剂可以在FECO大于95%的前提下达到工业级电流密度,约1 A/cm2.其优异的性能可归结于,富电子的Zn-N配位结构通过稳定*COOH中间体降低了反应能垒[59].这些研究在一定程度上阐明了配位数、价态对于催化性能的影响.除了Zn(Ⅱ)构型,Chen等也构建了一种Zn(I)-N3+1的结构,其中一个与锌配位的氮原子桥梁了两个石墨烯片层的边缘.原位衰减全反射-红外光谱(Attenuated Total Reflection-Infrared Spectroscopy,ATR-IR)谱图表明,扭曲的Zn-N3+1结构加快了CO2的活化和质子化过程(*CO2至*COOH),理论计算也表明,其活性位点上的反应势能与催化活性之间的构效关系,与之前的研究结论相类似.电化学性能表现为部分电流密度为50 mA/cm2,CO的法拉第效率达到了95%.
1.1.6 Ag和Au Ag是CO2RR的良好金属电极材料之一[61,62].早期被广泛研究的是块体Ag工作电极,直到近几年Ag 单原子催化剂才为人所知.王定胜和李亚栋等[63]报道了一种通过Ag 纳米颗粒和MnO2表面重构制备了一种Ag1/MnO2的单原子催化剂.环境透射电子显微镜(Environmental Transmission Electron Spectroscopy,ETEM),XRD 和DFT 计算系统地证明了Ag 纳米颗粒演变为Ag 单原子的过程.高温(400 ℃)诱导的MnO2的表面发生(211)至(310)的重构,(310)晶面的形成是稳定Ag 单原子的关键.催化剂在-0.85 V(vs.RHE)下达到了95.7%的FECO,-0.9 V 下达到了-6 mV/cm2.DFT 计算表明,Ag单原子位点具有较高的电子云密度,有助于促进电子从催化剂到CO2的转移.可以发现,Ag单原子催化剂所表现出的CO电流密度均较小,有待进一步改进.
与Ag单原子类似,Au单原子催化剂的报道也相对较少.最近的研究显示,在具有拉伸应变的Pd纳米颗粒上,负载的Au原子可以解决Pd的CO中毒问题[64],Au原子可以降低*CO的稳定性,而不影响CO2RR 中的其它中间体的稳定性.同时具有拉伸应变的Pd 表面可以稳定电还原过程中所有的中间体.与Pd/C 催化剂相比,在-0.25 V(vs.RHE)下,该催化剂表现出显著提升的CO 电流密度和质量活度.FECO达到了99%.
1.1.7 其它过渡金属 除了Fe,Co,Ni,Cu和Zn被广泛报道外,其它过渡金属也有少量报道.负载在超薄氮掺杂石墨烯上的Mo单原子可以催化CO2还原得到甲酸,产率可达747 mmol··h-1[65].Mn单原子的文献报道涉及调整配位原子种类,以及分子桥联策略.
1.2 主族金属单原子催化剂
1.2.1 p区金属 由于对氢析出反应(HER)有较高的过电位[8],铟(In)、锡(Sn)和铋(Bi)基等p区金属催化剂在电催化CO2还原领域中研究广泛.理论计算和实验均表明,对于非单原子催化剂而言,In[66~68],Sn[69~71],Sb[72],Bi[73]倾向于选择性生成HCOOH 或HCOO−.将金属(In[12,74],Sn[75],Bi[76,77])分散到单原子层级时,产物选择性通常也是如此.
与过渡金属单原子催化剂类似,M-N4也是p区金属单原子常见的存在形式.这些单原子催化剂通常表现出对C1产物具有较高的选择性.李亚栋和王定胜等[12]报道了具有Inδ+-N4结构的In 单原子催化剂.拓展X 射线吸收精细结构谱(Extended X-ray Absorption Fine Structure,EXAFS)显示,没有In-In 金属键的存在,In 原子第一配位壳层为4 个N 原子,In—N 键长为1.6 Å(1 Å=0.1 nm),符合In-N4结构.该催化剂表现出了对甲酸产物的高选择性,在-0.95 V(vs.RHE)时,法拉第效率为96%,TOF 达到12500 h-1.该工作是利用ZIF-8封装In(acac)3,经高温热解碳化,得到氮掺杂碳锚定In单原子.Lu等[78]采用了类似的方式,将金属盐更换为In2(SO4)3,所得催化剂达到了相近电位下转换频率几近翻倍的效果.这表明金属盐前驱体种类在一定程度上影响催化剂的活性.我们课题组采用了另外一种思路合成了In-N4单原子催化剂,最终得到了完全不同的C1产物.Zu等[79]通过一种快速的冷冻真空干燥后热解的策略,实现了带有正电荷的Sn 单原子(Snδ+)催化剂的合成.催化剂的结构和形貌通过XAFS 和HAADF-STEM 表征得到证明.原位傅里叶变换红外光谱表明,Snδ+活化CO2,通过稳定中间体CO·−2以及HCOO-*来实现后续的质子化过程.同时,氮掺杂也促进了决速步骤HCOO-的脱附过程.因此其HCOO-生成的起始电位仅为60 mV,TOFHCOO−达到了11930 h-1.其电还原活性可以保持200 h.本课题组通过金属-载体相互作用,合成了带有氧空位的Sn1/CuO复合单原子催化剂.Zhang等[76]通过热解Bi基MOF也可以获得Bi-N4的结构,在Bi纳米颗粒热原子化为Bi单原子的过程中,额外添加的双氰胺热分解产生的NH3捕获了Bi单原子,形成具有Bi-Nx结构单原子催化剂.其不同热解温度下材料形貌图以及不同材料的结构表征如图2(A)~(F)所示.图2(F)表明,BiSAs/NC 中Bi确以单原子分散的形式存在.该催化剂在较低过电位下获得了97%的CO 法拉第效率,TOF 达到了5525 h-1.DFT 计算表明,由于具有较低的反应能垒,Bi-N4结构促进了CO中间体*COOH 的快速生成.由氯化锑、活化的碳黑和尿素共热解制备而得的氮掺杂多孔碳负载的Sb单原子催化剂,实现了高效电还原CO2制备CO,TOFCO达到了16500 h-1[80].此外,另一种具有Sb-N4结构的单原子催化剂,其CO2还原至甲酸盐的选择性在-0.9 V(vs.RHE)达到了94%[81].DFT 计算表明,Sb-N4位点更有利于HCOO*的形成,因此产物主要为甲酸盐而不是CO.这与前文的Sb单原子催化剂报道的产物不同.可能归因于制备方法的不同,导致了单原子位点电子结构的变化,利于吸附的中间体由*COOH 变为HCOO*.催化剂制备方法上,氮掺杂碳上Sb-N4主要通过SbCl3与双氰胺、均苯三甲酸共热解得到.原位XAFS 和DFT 证实,Sbδ+-N4位点上发生CO2RR至HCOO-的有利途径.对于已报道的这些催化剂,其法拉第效率都达到了相对较高的水准,但电流密度还较小,远远达不到工业级应用的要求(1 A/cm2).
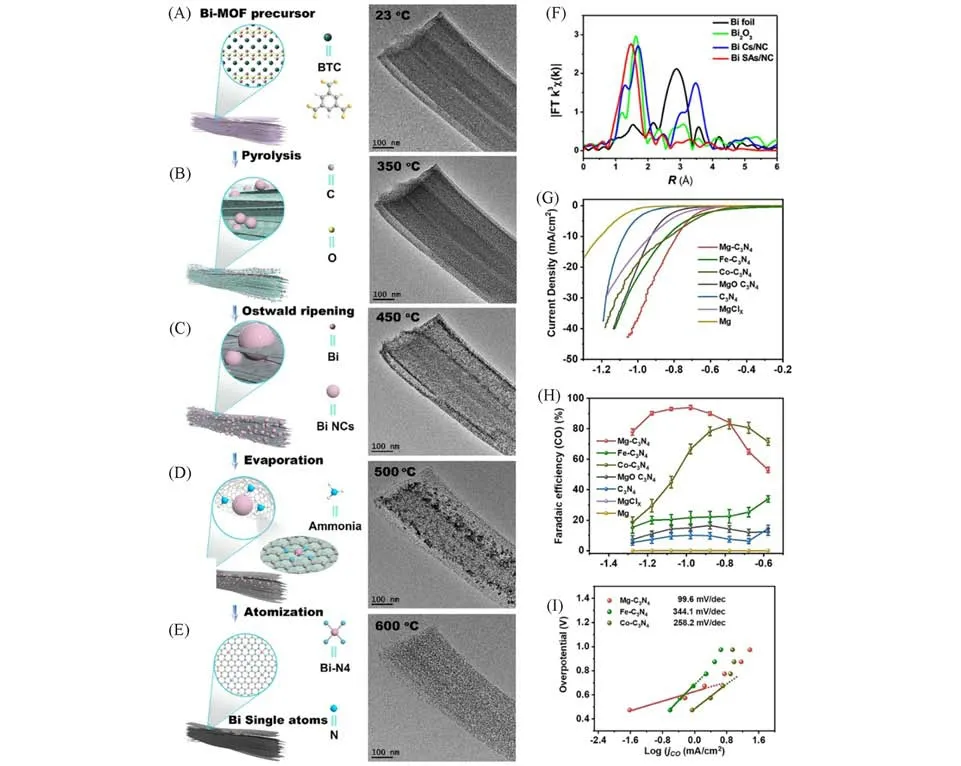
Fig.2 Scheme of the transformation from Bi⁃MOF to single Bi atoms and corresponding representative TEM images of Bi⁃MOF pyrolyzed at different temperatures with the assistance of DCD(dicyandi⁃amide) in situ(A—E), k3⁃weighted χ(k) function of the EXAFS spectra of Bi foil,Bi2O3,BiCs/NC,BiSAs/NC(F)[76],electrochemical CO2RR performance of LSV(Linear sweep voltammetry) curves at scan rate of 10 mV/s in H⁃cell(G),FECO at different potentials in H⁃cell(H)and Tafel plots(I)of Mg⁃C3N4[83]
1.2.2 s 区金属 与过渡族金属不同的是,s 区金属由于不具备激活的未占据d轨道,难以与CO2还原过程中的中间体形成特异性键合,通常表现为较差的CO2电催化还原性能,一直鲜有报道.但有研究人员指出,过渡金属由于其具有方向性的定域d轨道,会与CO 中的5σ和2π*轨道部分成键,导致与CO2电催化还原过程中的中间体(如*CO)的结合作用较强,因此中间体难以脱附生成产物或被进一步还原[53,82].主族金属的s轨道不具备方向性和定域性,这就导致其与CO的结合能力比较弱,有可能提高其CO脱附能力.作为植物光合作用活化CO2的关键金属元素,Mg原子具有3s轨道,单原子Mg可能具有活化CO2和优良的CO脱附能力.如,嵌入在石墨化C3N4中的Mg单原子表现出良好的CO2还原至CO性能[83],在H型电解池中,FECO在90%以上,且TOF约为18000 h-1.其CO2性能测试结果如图2(G)~(I)所示.进一步组装于气体扩散电极上时,可在FECO大于90%的条件下维持−300 mA/cm2的电流密度.DFT 算表明,在Mg-C3N4上形成*CO 的自由能分别比Fe-C3N4和Co-C3N4的高出1.24 和1.39 eV,揭示了其高CO选择性的原因.
2 单原子微环境的调控
调控单原子的微环境从而改变原单原子的本征催化特性,已成为当前研究的热点之一.非贵金属的单原子配位环境的调控可以进一步提高催化性能,甚至达到媲美贵金属催化剂的水平.这种方法规避了高成本的贵金属催化剂的使用,转而选择地球上含量丰富的金属元素,具有实现大规模工业化应用的潜力.
2.1 杂原子配位
2.1.1 改进合成方法 与金属原子配位的杂原子可以调控金属中心电荷密度,微调其与CO2电还原过程中中间体的结合强度,最终实现对某目标产物的高选择性、高产率合成[84].本课题组[8]采用了有机连接配体(1,3,5-苯三甲酸,H3BTC)和In(NO3)·5H2O合成In-MOF,后续通过与双氰胺物理混合后进行一步热解,得到了具有In-N4结构的InA/NC.控制实验和DFT计算表明,不同于另一研究的Inδ+-N4结构,InA/NC的In-N位点不仅有利于COOH*的解离生成CO,而且阻碍了甲酸盐的生成,使其对CO的选择性高于对甲酸盐的选择性.同时,由于In-N4的结构维持了催化界面附近碱性环境,也减弱了氢质子的还原.另外,以离子液体作为电解液,既增加了CO2底物的浓度又抑制了析氢副反应发生[8].这些因素共同促进了CO2还原至CO,在39.4 mA/cm2下表现出97.2%的CO 法拉第效率,并具有较高的TOF(约为40000 h-1),部分结果如图3(A)~(D)所示.这种调整单原子配位环境改变产物选择性的方法,为进一步探索单原子结构-性能关系奠定了基础.针对目前CO2RR的电流密度较低的问题,Chen等[85]提出了一种新颖的胺化策略,来提高Ni 单原子催化剂CO2RR 的电流密度.热解Ni 掺杂ZIF-8 形成的Ni-N4/C通过与尿素共热,再通过水热法与氨水反应实现了对Ni单原子催化剂的氨基化修饰,傅里叶转换红外光谱(Fourier Transform-Infrared Spectroscopy,FTIR)和XPS 均证明了—NH2的成功修饰.在流动池中测试发现,在−0.89 V 下FECO接近90%,且电流密度显著提升,达到了450 mA/cm2,可归结于修饰的—NH2调整了催化剂的电子结构,增强了*CO2和*COOH中间体的吸附和电荷转移效率.
2.1.2 调整配位原子数目和种类 对于Fe单原子催化剂,已有报道证明了在Fe位点引入额外的N配位原子能够促进CO2电还原效率.Cheng等[6]率先报道了含有Fe-N5位点的催化剂,通过电纺丝、惰性气氛下碳化和在氨气氛下氮化,构建了在嵌有Fe-N4位点的石墨片层中封装FexN纳米粒子的复合结构催化剂.该催化剂在-0.53 V(vs.RHE)下,实现了95%的CO 法拉第效率以及4.71 mA/cm2的分电流密度.FexN中N对Fe位点的配位作用,有效地解决了CO与Fe位点过度结合导致催化剂中毒的问题[31].具有明确Fe-N5位点的催化剂,是通过血红素、三聚氰胺和石墨烯进行共热解,并通过硫酸酸浸得到的.在不加入石墨烯的情况下,只能得到Fe-N4位点结构的单原子催化剂.在三聚氰胺热解的过程中,引入的石墨烯掺入了氮杂原子,分散在此载体上的Fe-N4位点将与额外N 原子发生轴向配位,得到了Fe-N5位点.基于此,该催化剂在过电位为0.35 V的条件下,FECO约为97%.理论计算表明,额外的轴向配位使得Fe3d轨道中的电子进一步减少,因此降低了Fe-CO之间的反馈π键,从而CO更容易从Fe位点上脱附[86].
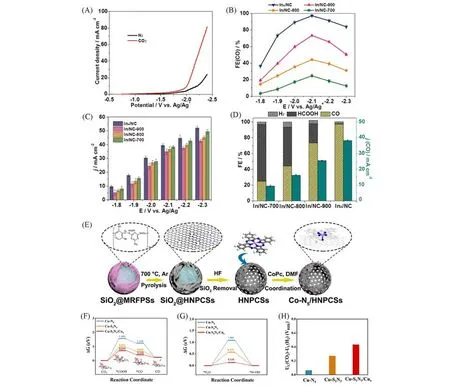
Fig.3 CO2 electroduction in 0.5 mol/L [Bmim]PF6/MeCN electrolyte(A—D)[8],schematic illustration of Co⁃N5/HNPCSs(E)[87],DFT investigations of Cu⁃S1N3/Cux,Cu⁃S1N3 and Cu⁃N4(F—H)[51]
Co-N位点也可通过引入额外N配位原子而增强CO2RR选择性.Pan,Lin和Chen等[87]通过模板法制备了中空氮掺杂碳小球,并通过浸渍吸附酞菁钴(CoPc),获得了具有Co-N5配位构型的电催化剂,中空氮掺杂碳小球中的氮原子作为额外氮源与CoPc中的Co-N4位点键合.其合成示意图如图3(E)所示.原位XANES 表征确定了催化活性位点为Co-N5.且催化剂在较宽的电势窗口下(-0.57~-0.88 V)保持着90%以上的CO 法拉第效率,在-0.73~-0.79 V 下FECO可大于99%.同时,10 h稳定性测试后,电流密度和法拉第效率几乎没有改变,也表明该催化剂具有良好的稳定性.理论计算进一步解释了Co-N5位点上形成*COOH的自由能较低,且CO脱附更为容易.通过对比Co-N4位点与Co-N4−x-Cx也能够发现类似的趋势.在-0.8 V(vs.RHE)下,前者FECO为82%,后者FECO为47%[88].另外,通过比较Ni-C/Ni-N的配位环境对电催化性能的影响,结果表明,配位微环境的变化会导致性能也随之改变,进一步证实了上述结论[89].值得注意的是,他们在早期研究配位数对催化性能影响的工作中呈现了截然不同的规律,随着N原子配位数的下降,CO的选择性提高.通过调节退火温度,得到不同配位数的Co-Nx,且随着温度的升高,x的数值也逐渐下降.这其中Co-N2展现出最突出的性能.在520 mV 过电位下达到了94%的CO法拉第效率,相应分电流密度为−18.1 mA/cm2.理论研究表明,Co原子附近N原子配位数的降低,产生了更多的未被填充的3d轨道,利于CO·−2的吸附,同时Co-N2具有最低的电荷转移阻抗,即促进了CO2活化,加速了电荷速率,导致了CO2RR 性能提升[90].显然,这两个研究中N 原子与中心Co原子配位的方式是不同的,前者增加了轴向配位,后者减少了平面内的配位,并且减少的N 原子由C原子替代,这些结果进一步说明,改变配位原子的配位方式将显著影响催化性能.
同样,通过改变热解温度,Gong和Jiao等[91]合成了一系列NiSA-N4−2-C催化剂.其中,NiSA-N2-C相比于NiSA-N3-C 和NiSA-N3-C 催化性能更优.其最高的FECO达到了98%,TOFCO为1622 h-1.理论计算揭示,NiSA-N2-C更有利于*COOH的形成.另有实验表明,通过等离子辅助法和氮空位诱导配位重构的策略成功得到了具有Ni-N2位点的催化剂,得益于其特殊的电子结构,CO2分子吸附并活化的自由能垒更低[92],因此该催化剂能够在较小的过电位(590 mV)下实现96%的FECO,并在890 mV 过电位下电流密度达到33 mA/cm2.此外,考虑电荷容量和氢键相互作用,理论计算进一步指出,Ni-N1C3与其相应的系列(Ni-NxC4−x)相比,活性和选择性更高[93].
近期,Zheng等[94]报道了一种等离子体激活策略合成Cu基MOF单原子催化剂.该催化剂中预先与Cu 配位的氧原子通过等离子体处理而被部分去除,因而形成了富有氧空位的低配位Cu 单原子位点.同时,等离子体处理也导致了材料产生多孔的特性.低配位Cu 单原子和分级多孔的结构共同促进了CO2RR.电化学性能显示,CH4的最大法拉第效率达到了75.3%.二氧化碳还原产物的选择性达到了96.5%.相应的电流密度为47.8 mA/cm2.
除了上文所描述N配位原子数减少后产生的空位被C原子取代,也有文献报道了Ni-NxV4-x构型的催化剂(其中V代表缺陷,即无配位原子)可以抑制析氢反应发生[22].这种含有配位不饱和的Ni单原子催化剂通过热解含Ni 的ZIF-8得到,Ni的质量负载量达到了5.44%,高于多数文献报道.该催化剂在-1.03 V(vs.RHE)下达到了(71.5±2.9)mA/cm2的电流密度,且在-0.53~-1.03 V的电势范围内,维持92.0%~98%的CO 法拉第效率.另一研究展现了Ni-N3位点对于CO2RR 生成CO 的促进作用.其CO 法拉第效率超过了90%,转换频率达到12000 h-1,质量活度为10600 mA/mg[95].Rong 和Wang 等[75]基于Ni-O之间的弱相互作用,直接热解含有NiII-N/O的前体,随着O的移除产生空位V,就可得到Ni-N3-V的结构.该催化剂表现出远优于Ni-N4单原子的性能,TOF为1.35×105h-1,大于Ni-N4的4倍.综上所述,这些结果都证明了Ni-N(S)xV4−x构型的优势.
除了配位数之外,改变配位原子种类是调控单原子电催化活性的另一种策略.引入电负性较小的原子可以很好地降低CO2RR 过程中间体的能垒,并最终加快反应的动力学.在碳基平面上,如图3(F)~(H)所示,不对称的Cu-S1N3原子界面相比于Cu-N4位点具有优化的*COOH结合能,另外,附近的Cu纳米颗粒(Cux)促进了质子化过程.因此Cu-S1N3/Cux在-0.65 V下表现出近100%的FECO,-0.55~-0.75 V 区间内维持90%以上的FECO,优于Cu-N4(-0.7 V 时,FECO=54%)和Cu-S1N3(-0.7 V 时,FECO=70%)[51].Zhang 等[96]通过MOF 节点联结单位点Cu 得到的具有Cu-O4位点催化剂(Cu-DBC),其甲烷选择性接近80%,在-0.9 V(vs.RHE)下,部分电流密度达到−203 mA/cm2.相同条件下,Cu-O4位点比Cu-N4位点具有更低的CO2RR反应能垒.S掺杂NiN2得到的NiN2-S,在高的过电位施加的条件下会产生S空位,而S原子和S空位都具有降低CO2生成CO的反应能垒的作用.因此在-0.8 V(vs.RHE)下,该催化剂FECO达到97%,-0.9 V(vs.RHE)下CO分电流密度为40.3 mA/cm2[97].此外,改变配位杂原子种类也能影响反应路径,得到不同的产物.如SnN3O1表现出独特的性质,与Sn-N4的主要产物为HCOOH和H2相比,其主要产物为CO.理论计算表明,SnN3O1构型能够降低*COO活化和*COOH形成的能垒[98].Cd-N4S1中S 原子与Cd 原子轴向配位,同时Cd 原子锚定在Cd-N4的石墨碳平面上[99].其结构示意图如图4(A)所示.这种轴向的S原子配位不仅进一步降低了CO2还原反应能垒,同时也抑制了析氢反应.得益于此,Cd-N4S1表现出优越的CO2RR性能,在H型电解池中达到182.2 mA/cm2的CO分电流密度和超过95%的法拉第效率,转化频率可达73000 h-1,是目前在H型电解池中报道的最优性能之一.理论计算表明,Cd-N4S1单原子相比Cd-N4就具有降低CO2反应能垒的作用.通过化学气相沉积法,将Sn-Ox单核物种锚定在碳化的ZIF-8中.然后进一步借助聚四氟乙烯热解生成的C2F4刻蚀ZIF-8来实现氟化,最终合成了具有Sn-C2O2F 结构的单原子电催化剂.其在较宽的电位内(-0.2~-0.6 V)可以保持大于90%的FECO,最大可达95.2%.而相同条件下,Sn-N4以甲酸盐作为主要产物.通过C,O原子配位调控实现了对中间体吸附的优化,而轴向的F原子引入抑制了析氢副反应,促进了CO2到CO的转换[100].但如图4(B)所示,抑制析氢反应的同时,相比Sn-C2O2,也一定程度上增加了*COOH+H+的形成能垒.基于此,该催化剂在490 mV 的过电位下,FECO为90.5%,电流密度为10.8 mA/cm2,转换频率达到1566 h-1.Zhang等[101]通过原位XAS和DFT计算阐明,轴向的Cl配位原子引入能够有效地降低Mn-N4位点上关键中间体的形成能垒.其结构表征结果如图4(C)和(D)所示.在0.49 V的过电位下,CO电流密度约为10 mA/cm2,最大法拉第效率为97%.上述研究表明,轴向配位杂原子的引入对不同的金属单原子位点展现出不同的性质.另外,Chen等[102]通过熔融盐辅助策略,快速热解和可控活化的策略合成了Fe1N4-O1催化剂.虽然其氧原子也是处于轴向配位的位置,但这个氧原子处于另外一个石墨碳平面内,并不同于前者的独立于面外放的氧原子.这种氧原子可以使得电子定域化,促进CO的脱附,同时提升析氢反应的能垒.因而使得催化剂在-0.56~-0.87 V下获得接近100%的FECO.不与金属直接配位成键的杂原子也会影响金属单原子的电子结构.如在第三配位壳层中,以P—C键为成键形式的P原子会增强Fe单原子的电子定域化[103],从而增强了其*COOH中间体的稳定性,在较低的过电位(320 mV)下表现出97%的CO选择性.
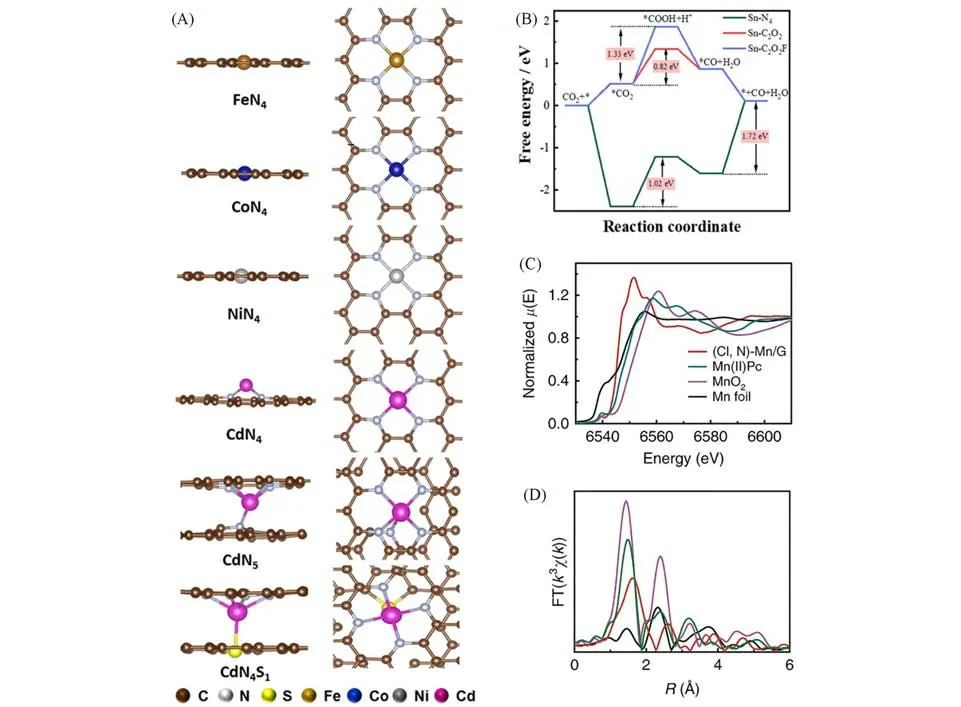
Fig.4 Side and top view of the FeN4,CoN4,NiN4,CdN4,CdN5,and CdN4S1 models(A)[99],Gibbs free energy diagrams for formate formation on Sn⁃N4,Sn⁃C2O2,and Sn⁃C2O2F(B)[100],XANES(C)and EXAFS(D)spectra at Mn K⁃edge[101]
综上所述,绝大部分研究主要集中于通过精细调控金属单原子的配位数、配位杂原子的种类以及配位结构以优化局部电子结构,调节中间体的吸附和结合强度,实现目标产物的高选择性,对于该策略对单原子催化剂整体活性的影响,还需要进一步研究.
2.2 双金属单原子
2.2.1 双金属中心 由于单原子活性位点单一,同时因负载量较低,原子与原子之间距离较远,不利于需要多电子参与的反应,且其对需要多步电子转移质子耦合和涉及众多反应中间体参与的化学反应(如二氧化碳还原到多碳产物)没有明显优势.此时,若存在两个相同的或者不同的金属单原子构成的双活性位点催化剂能够发挥协同效应的作用[104].Fe-N4对*COOH 的适中吸附强度导致其对CO2电还原的过电位很低,但从*COOH 解离而来的*CO 难以脱附[30].为了打破这个中间体的吸附能比例关系(Scaling Relationship),Lin 等[105]开发了一种Fe-N 位点和CoPc 构成的具有协同效应的催化剂,相对于Fe-N-C,CoPc@Fe-N-C能够在更宽的电势窗口下保持约90%的法拉第效率,且起始电位更低[−0.13 V(vs.RHE)].理论计算表明,CoPc 促进了Fe-N-C位点上*CO 的脱附,并抑制析氢反应,CoPc 的引入几乎不会影响*COOH的形成,与实验结果相吻合.
串联催化剂是一种通过组合两种不同催化组分的材料来改变催化性质的催化剂,由于不同的活性位点具有不同的性质,可以实现CO2多电子(>2 e)还原产物的目标[51,106],在单原子领域,CoPc@Zn-N-C 串联催化剂可以实现远高于单一组分的CoPc 或Zn-N-C 催化剂的性能[107],其CH4/CO 的产生比例大于单一组分催化剂.理论计算揭示了CO 先在CoPc 上生成,同时CoPc 位点还会促进*H 转移到Zn-N-C,然后CO扩散到Zn-N-C位点上被还原为CH4,其具体的DFT计算所揭示的结果如图5(A)和(B)所示.可见,在CoPc上,*COOH更容易形成.而Zn-N4更倾向于将CO转换为CH4.
2.2.2 双金属位点 与双金属中心不同的是,双金属位点通常由处于同一石墨碳平面中的两个金属原子所构成的活性位点,并对CO2还原过程具有协同催化的作用.理论计算结果显示,Cu/Mn,Ni/Mn,Ni/Fe 3种组合有可能打破*COOH和*CO之间的吸附自由能型比例关系[108].相关实验研究进一步验证了理论计算结果的正确性.Ni-Fe-N6-C 在-0.5~-0.9 V(vs.RHE)内实现了90%的法拉第效率[109].在-0.7 V时,FECO达到了98%,且30 h稳定性测试后,产物选择性几乎没有改变.根据理论计算的结果,Ni-Fe-N6-C具有更低的*COOH的形成能和*CO的脱附能.其中,非键合的Ni位点和Fe位点之间的协同作用也促进了*COOH 的形成.然而,该催化剂不影响*CO 脱附能垒,这也导致了其CO 电流密度较小[110].此外,Ni和Fe之间能够通过成键改变原子轨道能级,独特的电子结构和更高的Fe氧化态,减弱了反应中间体结合能,因此促进了CO2RR 的性能.690 mV 过电位下该催化剂具有50.4 mA/cm2的CO分电流密度,FECO为94.4%[111].此外,通过理论计算表明,N4-Ni-Sn-N4可以降低*OCHO的形成能垒,表现出优异的催化性能,甲酸产率为36.7 mol·h-1·g−1Sn,相应的TOF为4752 h-1[112],其结构和电荷示意图如图5(C)所示.Liang等[113]首次引入了稀土金属单原子La与Zn单原子组成双金属单原子催化剂.通过调整La和Zn之间的摩尔比,实现了不同摩尔比的CO/H2(合成气)的生成.
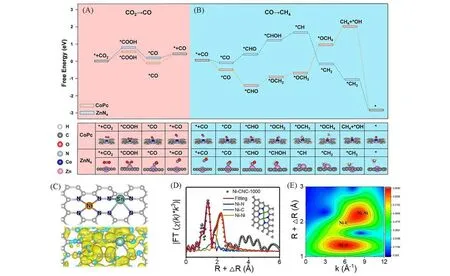
Fig.5 Free⁃energy profile and optimized configurations of intermediates in CO2electroreduction to CO(A),CO electroreduction to CH4 over CoPc and ZnN4(B)[107],a proposed model with spin electron density of NiSn⁃APC(yellow stands for spin up and green stands for spin down)(C)[112],corresponding EXAFS R space⁃fitting curves(D)and WT⁃EXAFS plot for Ni⁃CNC⁃1000(E)[116]
双金属位点催化剂中,用于CO2还原的同核双位点催化剂也有报道.李亚栋和陈晨等[114]将Cu 单原子对负载于Pd10Te3合金上,在Cu 原子对中,倾向于吸附CO2分子,而倾向于吸附H2O分子,共同作用促进了对CO2分子的活化,在−0.78 V左右,CO选择性达到了92%.这正体现出单原子所不具备的“两原子活化两种分子”的性质.此外,Ding,Liu和Tao等[115]通过热解一种具有邻近Ni原子占据的金属有机配合物,构建了一种通过两个氮原子连接的双Ni1-N4位点,理论计算表明,电催化测试条件下,氧原子桥联的两个Ni1-N4位点显著降低了CO2活化能垒,表现出较高的CO的选择性.Cao等[116]运用一种电纺丝-热解技术制备了Ni2-N4-C2位点的催化剂,其结构表征结果如图5(D)和(E)所示,印证了其结果.实验结果和理论计算均表明,这种与N,C原子键合的双核Ni双原子催化剂调整了d带的电子特征,优化了CO2及其中间体的吸附,增强了电荷和物质传输,促进了CO2还原.
目前,双金属单原子催化剂主要集中在C1产物上,仅有一部分串联催化剂实现了C2+产物的生成.结合双金属单原子催化剂具有不同活性位点的优势,实现C2+产物的生成是发展双位点单原子催化剂的内在原因之一.
2.3 金属-载体的相互作用
载体与金属单原子之间通过共价或离子键连接,从而调控金属原子的电子结构,实现金属单原子构效关系的改变[84,117].Cu 单原子位点与C3N4上C 位点分子内的协同作用促进包括C2H4,C2H6和C2H5OH在内的C2+产物的生成.Cu单原子位点是吸附*COOH,*CO和*CHO的活性位点.而临近C3N4上的C 位点则是*OCH2和*OCH3的活性位点[118].Sn 单原子位点、氧空位和CuO 基底之间的协调使得催化剂材料具备更高的双电层电容,更高的CO2吸附能力和更低的界面电荷转移阻抗[4].系统的实验手段和理论计算证明,Sn1/VO-CuO-90通过降低*COOH解离的能垒来促进*CO的形成.随后吸附到CuO基底上将*CO 进一步还原为甲醇.HAADF-STEM 和XAFS 进一步确认了Sn1/VO-CuO 的结构.电化学实验结果显示,该催化剂在67 mA/cm2的电流密度下达到了88.6%的甲醇法拉第效率.其在不同电位下的FEmethanol和j结果如图6(A)和(B)所示.在-2.0 V(vs.Ag/Ag+)的电位下,甲醇的法拉第效率达到了最大.从-1.7~-2.2 V(vs.Ag/Ag+)的电位下,j逐渐增大.还有研究表明,Sn单原子与富含氧空位的CuO可以协同促进C2H4的生成,理论计算表明,通过在CuO上掺杂Sn降低了*CO中间体之间C-C偶联的自由能,从而促进C2H4生成[119].负载于α-Co(OH)2基底上的Ir单原子可以更加有效地稳定吸附的CO2物种,同时促进了电子的快速传输从而生成CO[120].Cu 纳米颗粒上负载的Sn 单原子也表现出类似的性质,-1.0 V 时达到最大的FECO(95.3%)[121].单原子Pb 掺杂的铜基底调整了铜的电子结构,并由此将CO2RR的第一步质子化过程从到生成*COOH路径调整到生成HCOO*.最终实现了在工业级电流密度下(1 A/cm2)达到96%的甲酸法拉第效率.需要指出的是,该催化活性位点是Cu基底而非Pb单原子[122].以上结果均表明,金属-载体相互作用是一种优化单原子催化剂性能极具潜力的策略.
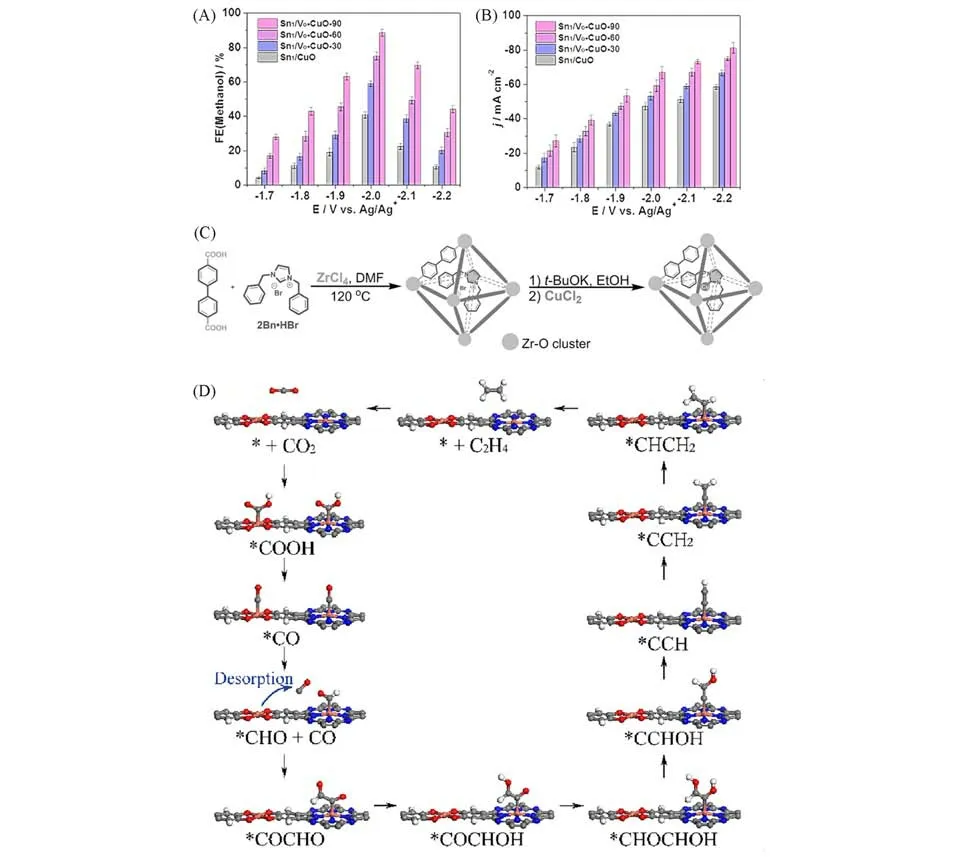
Fig.6 CO2 electroreduction in [Bmim]BF4/H2O electrolyte with a 1:3(molar ratio)on Sn1/VO⁃CuO⁃90,Sn1/VO⁃CuO⁃60,Sn1/VO⁃CuO⁃30 and Sn1/VO(A,B)[4],representation of the synthesis of 2Bn⁃Cu@UiO⁃67(C)[123],proposed CO2RR mechanism of PcCu⁃Cu⁃O(D)[126]
2.4 空间限域
空间限域策略可以通过创建微小的反应空间,促进CO2的多电子还原.近期,一种含有氮杂环卡宾配位的铜单原子位点的催化剂(2Bn-Cu@UiO-67)实现了前所未有的CH3OH 转换频率(TOFCH3OH=58680 h-1),同时在-1.5 V(vs.RHE)下达到了81%的甲烷法拉第效率,电流密度为420 mA/cm2,在宽的电势范围内仍可以保持优异的性能[123],其合成示意图如图6(C)所示.由于氮杂环卡宾配体对Cu单原子位点的给电子作用,增大了Cu位点的金属表面电荷密度,因此很大程度上优化了*CHO中间体的吸附,而*CHO是在CH4形成过程中的一个关键中间体.同时,UiO@67具有的孔道结构也具有较强的CO2捕获能力,孔道之间这种狭小的空间有利于CO2还原过程中产生的中间体从一个2Bn-Cu位点脱附后扩散到另一个2Bn-Cu位点,实现中间体的进一步还原,从而有利于CH4的产生.氮杂环卡宾配体的给电子作用,以及UiO-67在空间上提供的互连反应微空间协同催化了CO2电还原.该研究显示了空间策略具有的广阔前景.
2.5 分子桥联
共价有机框架(Covalent Organic Frameworks,COFs)能够将单金属位点分开,是构建单分散的金属单原子催化剂的理想模型之一.这种方式避免了传统方法需要的高温热解,可以在较为温和的温度下实现单原子位点的分散.如,常见的方式有直接利用酞菁配体上的4个氮原子与金属离子配位,构建金属酞菁结构单元,再通过共价有机化学反应实现结构单元之间的连接,获得具有超大共轭体系的金属-共价有机物网络结构.在锚定Mn单位点的COFbpy催化剂(COFbpyMn)具有明确可辨的配位中心,在水中还原CO2具有较低的过电位(190 mV),并在550 mV的过电位下电流密度达到了12 mA/cm2.TOFCO和TONCO(Turnover number,转换数)分别为1100和5800 h-1(在16 h后)[124],几乎是等量Mn基分子催化剂的10倍.Lu,Zhang和Liu等[125]利用二噁英连接金属酞菁实现了系列单原子位点催化剂的构建,NiPc-TFPN 和CoPc-TFPN 分别实现了(99.8±1.24)%和(96.1±1.25)%的FECO.此外,利用COFs 构建的含有Cu-N4和Cu-O4位点的催化剂(PcCu-Cu-O)也实现了CO2到多碳产物的高选择性,在0.1 mol/L KHCO3溶液中,-1.2 V(vs.RHE)测试条件下乙烯选择性达到50%.电流密度为7.3 mA/cm2.原位红外光谱实验也进一步验证了CuPc 中的Cu-N4位点和Cu-O4位点之间的协同作用[126],如图6(D)所示.Cu-O4位点脱附的CO可以与Cu-N4吸附的*CO发生C-C偶联反应.金属酞菁构建的MOF材料也表现出优越的性能.二维金属酞菁基MOF 纳米片(NiPc-NiO4)克服了传统MOF 导电性较弱的缺点[127],展现出98.4%的CO选择性,CO电流密度达到了34.5 mA/cm2.金属卟啉所组成的COF实现了多电子还原产物的生成.通过使用功能化剥离试剂对较大的COF 进行修饰和剥离,生成具有较大尺寸(约1.0 μm)并且超薄(约3.8 nm)的Cu-Tph-COF-Dct,展现出约为80%,在-0.9 V下,流动池中具有−220.0 mA/cm2的电流密度的性能[128].这几乎是未剥离的COF的两倍.DFT计算揭示,增强的性能归结于固定在了COF上,整合了氨基和三嗪基团功能化剥离试剂增强了CO2的吸附和活化,稳定了反应的中间体并增大了Cu位点附近的CO浓度.对于金属卟啉的MOF材料,将多层的二维卟啉基MOF制备成单层,也有利于C2产物的生成[129],这是由于单层的卟啉基MOF具有重构的Cu-O4位点,而多于双层的主产物主要为CO与HCOO-.但是,在仅施加一定电位的情况下,只达到了11.9%的,在添加了光的照射后,C2的法拉第效率提升到了41.1%.
3 总结与展望
综合评述了近年来单原子催化剂在电催化二氧化碳还原领域的研究进展,总结了包括过渡金属单原子、主族金属单原子在内的不同单原子种类,归纳提出了5种金属单原子催化剂的设计策略(包括杂原子配位、双单原子位点、金属-载体相互作用、空间限域和分子桥联),来调控金属单原子催化剂的微环境,以实现电催化CO2RR的高活性和高选择性.目前单原子催化剂(包括双金属单原子催化剂)的主要产物为CO,CO的选择性和电流密度能够在较宽的电位窗口内保持较高的水平.但是,如何进一步提升催化剂的性能使之能够在工业条件下长期稳定运行仍然是目前需要解决的问题.另外,对于生成多电子(>2e)还原产物以及C2+产物,目前的单一活性组分的单原子催化剂难以达到.这是因为,单原子催化剂一般金属负载量较低(<5%),活性位点之间距离较远,CO2还原后生成的CO从活性位点脱附后较难扩散到另一个单原子位点,碳-碳偶联发生的可能性较低,从而导致了以CO,HCOOH和CH4等为主导的C1产物.将金属单原子引入到串联催化剂或含多组分的多位点催化剂中是有望生成多电子产物以及C2+产物的策略之一.如何通过实验手段系统而精确地调控金属单原子的局部微环境,仍需进一步探究.此外,对于单原子催化剂上发生的CO2电催化还原反应机理的深入研究也极具挑战性,明晰反应机理可有的放矢地设计单原子催化剂的构型,从而实现更高的催化活性和选择性.未来,可以期待借助明确定义的单原子催化剂催化活性位点,结合系统的准原位以及原位的表征方法,来深入探究CO2电催化反应的机理和路径,并通过理论计算模拟进行辅助指导验证,从而最大程度地将材料结构、催化活性和反应机理有机统一起来.
