Practical insights into chronic management of hepatic Wilson’s disease
2022-06-23EricaNicolaLynchClaudiaCampaniTommasoInnocentiGabrieleDragoniPaoloForteAndreaGalli
lNTRODUCTlON
Wilson’s disease (WD) is a rare autosomal recessive disease which leads to the accumulation of copper. WD is due to defective mutations of the ATP7B gene, which encodes an ATPase that is responsible for the biliary excretion of copper and its incorporation into apo-ceruloplasmin to form holo-ceruloplasmin. Holo-ceruloplasmin is the enzymatically active form which has catalytic and antioxidant activity and delivers copper ions to the organism[1]. In healthy individuals, the bile is the main route of elimination of copper[1]. In WD patients, copper accumulates in the liver and is released into the bloodstream, where it circulates loosely bound to albumin, transcuprein, and low molar mass molecules[2]. Finally, it is excreted in urine. Copper can also be accumulated in the brain, kidneys, heart, and osseous matter and causes damage due to direct toxicity or oxidative stress[3]. WD mainly has hepatic, neurologic, psychiatric, or mixed manifestations.
PREVALENCE AND CLlNlCAL MANlFESTATlONS
In 2020, Sandahl
[4] analysed recent literature and results based on different methodologies and produced an updated estimate of the prevalence of WD disease of 1:30000-1:50000, which appeared to be valid for United States, Europe, and Asia; their findings were surprisingly close to the “Scheinberg-Sternlieb Estimate” proposed in 1984. The estimated prevalence is higher in some particular regions due to increased frequencies of the disease-causing mutations and/or more first-cousin parenthoods (
, Sardinia, Italy, 1:8700)[4]. In a recent study conducted in Greece, 57.1% of WD patients were asymptomatic at diagnosis (with only mildly elevated liver enzymes), 20.6% of patients suffered from neurological disease, 12.7% had an overt liver disease, 6.3% of patients suffered from acute liver failure, and 3.2% had other manifestations[5]. Specifically, 36.5% of patients were already cirrhotic at time of diagnosis[5]. The most common neurological manifestations are dysarthria (91%), gait disturbance (75%), risus sardonicus (72%), dystonia (69%), rigidity (66%), tremor (60%), and dysphagia (50%)[6]. Approximately 50% of patients with neurologic symptoms have liver cirrhosis at the time of diagnosis. It is unclear whether there are WD patients with neurologic symptoms without any liver involvement. WD can present at any age, although it is most frequently diagnosed in patients between the ages of 5 and 35, with neurologic patients being generally older than those with hepatic manifestations[7].
Asymptomatic patients with WD
Family screening for WD with ATP7B gene mutational analysis has led to an increase in the identification of asymptomatic siblings with homozygous or compound heterozygous pathological mutations for WD. The most recognised diagnostic score for WD, the Leipzig scoring system, allows the diagnosis of WD on the basis of compatible mutations on both chromosomes, without requiring altered copper metabolism tests[7]. In most cases, asymptomatic siblings with a positive genetic test are considered to have mild liver disease and are started on treatment for WD. The disease is considered fully penetrant, so, according to current guidelines, its genetic diagnosis is a mandatory indication for copper-depleting treatment, although not all gene changes have been established as causing WD[7-9].
Członkowska
[8] reported the case of an 84-year-old woman, who was diagnosed with WD at the age of 54 years and refused anti-copper treatment. The only clinical manifestation of her disease were Keiser-Fleischer rings. Her ceruloplasmin levels were normal and urinary copper excretion (UCE) was slightly elevated (90 μg/24 h, normal values below 50 μg/24 h). Stättermayer
[10] suggest that asymptomatic siblings diagnosed by genetic screening require further testing before initiating treatment, as they reported two patients carrying two disease—causing mutations who did not have any evidence of altered copper metabolism. In a French study by Collet
[11], 697 indiscriminate subjects were tested for WD mutations: a considerable discrepancy between the heterozygous carrier frequency and the clinical prevalence of WD was found which could be explained by incomplete penetrance or to modifiers genes. On the other hand, it should be noted that there are a few reported cases of presymptomatic siblings with only mild biochemical alterations, but with histological evidence of advanced liver disease at diagnosis[12]. Hence, the Leipzig scoring system could be modified in the future as genetic testing might not be considered sufficient to diagnose WD, although further studies are needed to better understand the complex pathophysiology and clinical variability of WD.
Finally, there is knowing. I know Scott will throw his laundry just shy of the hamper14 every night; he ll be late to most appointments and eat the last chocolate in the box. He knows that I sleep with a pillow over my head; I ll lock us out of the house at a regular basis, and I will also eat the last chocolate.
Perhaps one of the enduring elements of the Cinderella story comes from the politics of a family, usually a blended family. While many fairy tales have outside antagonists50, Cinderella s trials are in her home and immediate23 family. Return to place in story.
TREATMENT
D-Penicillamine
D-penicillamine (DPA) is the first oral copper chelating agent which was approved for the treatment of WD in 1956[13]. Until then, the only available chelating agent was dimercaprol, a parenterally administered drug still used for the treatment of arsenic, gold, copper, and mercury poisoning, as well as for severe symptomatic WD[14]. DPA binds to extracellular copper and promotes its urinary excretion. Moreover, DPA may also induce the release of intracellular stores of metallothionein, an endogenous copper chelator[15,16]. DPA should be administered at least 1 h before or 2 h after meals, as food inhibits its absorption by almost fifty percent[7]. DPA should be started at a daily dose of 250-500 mg It should be gradually increased by 250 mg/d every 4-7 d up to 1000-1500 mg/d (2-4 divided doses) to reduce the risk of adverse events (AE) and of paradoxical worsening in patients with predominantly neurologic symptoms[9]. The daily dose can be reduced to 750-1000 mg (2 divided doses) during maintenance[9]. DPA can be also used in children at a dose of 20 mg/kg/d[9]. DPA is associated with numerous AE, which cause treatment discontinuation in approximately 30% of patients[17,18]. Early AE (during the first 3 wk of treatment) include fever, generalized pruritus and rashes, lymphadenopathy, and proteinuria[9]. Moreover, the risk of deterioration of neurologic manifestations increases during the first weeks of treatment in patients with neurologic manifestations of WD and worsening of neurological symptoms has been reported in up to 50% of patients[9,19,20]. Litwin
[21] showed that in 143 symptomatic WD patients a neurological worsening at the beginning of anti-copper therapy occurred in over 10% of patients. The Authors stated that special attention should be paid to those with severe initial neurological manifestations, advanced brain injury, and to those on therapy with dopamine receptor antagonists. In a more recent study by Zhang
[22], 33.8% of patients exhibited significant neurological deterioration after the first 4-weeks of DPA monotherapy. The underlying mechanism causing neurological worsening at treatment initiation seems to be related to a rapid mobilization of unbound copper which triggers a cytotoxic effect in neuronal tissue[23]. Late AE include progressive proteinuria, gastric symptoms, hair loss, hypogeusia, leukopenia, thrombocytopenia, or total aplasia due to bone marrow depression, and myasthenia- or lupus-like syndrome marked by haematuria and positive antinuclear antibody[24]. Finally, elastosis perforans serpiginosa, pemphigus or pemphigoid-like lesions, lichen planus, and aphthous stomatitis have been reported[25].
They could fly over the sea in ships, and mount the high hills which were far above the clouds; and the lands they possessed66, their woods and their fields, stretched far away beyond the reach of her sight
Trientine
Trientine has been approved as oral copper-chelating agent in 1969. It leads to a hepatic improvement in more than ninety percent of patients with WD mainly affecting the liver[26]. Trientine increases UCE similarly to DPA. In some countries it is only available as a second-line treatment. Trientine gastrointestinal absorption appears to be reduced if compared with DPA, and therefore trientine should be assumed 1 h before or 3 h after meals[7,9]. The initial daily dose of trientine is 20 mg/kg (in 2-4 divided doses), which can be reduced to 15 mg/kg during follow-up. There is no established weightbased dose for the paediatric population and most of the experts use the dose of 20 mg/kg/d with similar dosing frequency as in adults[9]. Trientine is associated with fewer side effects compared with DPA. First of all, the paradoxical neurological worsening at treatment initiation was believed to be less frequent[9,27], although more recent retrospective studies have observed a comparable incidence of paradoxical worsening in patients treated with DPA, trientine, and zinc salts[20,26]. Secondly, bone marrow suppression with thrombocytopenia and leukopenia is rare in patients on trientine, and it should prompt evaluation to identify possible copper deficiency from the overtreatment[23]. Moreover, a reduced incidence of lupus-like syndrome compared with DPA has been described[9]. AE related to trientine are generally mild and include headache, arthralgias, myalgias, nausea, anorexia, diarrhoea, rash, and renal dysfunction[9].
Everyone would call him a tyrant33 if he were to give such an order--in fact, he dared not try it!At length he collected himself enough to say:-- If this young man will enlist34 in my army I will let him off
Zinc salts
Ah! Ivan, Ivan, exclaimed Marie, trembling with joy, heaven has sent us a child at last! And she threw herself upon Snowflake (for that was the snow-child s name) and covered her with kisses
Potential new pharmacological treatments
New treatment options are being studied mainly to reduce the risk of neurological deterioration during treatment or to improve patient compliance. The FOCUS phase III study comparing bis-choline tetrathiomolybdate (TTM) with other standard-of-care anti-copper compounds has been started in 2018. TTM binds to copper to form an inert protein complex that cannot redistribute to the central nervous system and for this reason could be a future treatment option for neurological patients[38]. Sodium 2,3-dimercapto-1-propane sulfonate (DMPS) is an intravenous copper-chelating agent that forms complexes with copper which are excreted by the. In China, DMPS is routinely employed for the treatment of WD. In combination with zinc, it was found to be more effective than zinc alone for the treatment of patients with a history of neurological deterioration during DPA[39]. In a recent retrospective study on 158 patients, the combination of DMPS and zinc was also found to have better results than DPA in neurological WD[22]. To improve adherence in the long term, the possibility of administering a single daily dose of trientine as a maintenance therapy has been explored in a study that included 8 patients who were followed for 12 mo[40,41]. The once daily dosing of trientine seems to improve compliance while maintaining effectiveness[40,41]. Larger trials with longer duration of follow up and testing of dose response are required to assess the safety, long-term efficacy and cost effectiveness of single dose trientine treatment for maintenance therapy in WD. Gene therapy approaches for replacement of the mutated ATP7B have been successful in animal models of WD and remain a potential mean to cure WD[42].
A recent multicentre, randomized, non-inferiority, open label study has evaluated the efficacy and safety of trientine tetrahydrochloride
DPA in patients with stable WD. Fifty-three adult patients with clinically stable disease for more than one year were followed for a 12-week reference period before being randomized 1:1 to trientine tetrahydrochloride or DPA twice daily. The primary endpoint of the study was to obtain non-ceruloplasmin-bound copper (NCC) levels within the therapeutic range. NCC was assessed with a novel method based on copper speciation. The secondary endpoint was to obtain both NCC and UCE rates within the optimal range. The CHELATE study met its primary efficacy endpoint by demonstrating that trientine tetrahydrochloride was non-inferior to DPA. Five serious AE were observed in the 27 patients treated with DPA, while none were observed in the 26 patients randomized to trientine tetrahydrochloride [NCT03539952]. Trientine tetrahydrochloride is produced as a scored tablet which is stable at room temperature, in contrast with trientine dihydrochloride which is a capsule and needs to be stored in the fridge[28,29].
Vitamin B6
DPA therapy has been associated with biochemical evidence of pyridoxine deficiency. In the literature, only one case of clinically relevant pyridoxine deficiency induced by DPA has been reported. A man, whose nutritional status and therapy regimen were not specified, developed peripheral sensory neuropathy (WD does not affect the sensory nervous system) shortly after having initiated treatment with DPA. The symptoms rapidly cleared with vitamin B6 integration, without DPA discontinuation[43-46]. Epilepsy was initially suggested to be a manifestation of vitamin B6 deficiency in WD patients treated with DPA, but this finding was not confirmed in later studies[47,48]. Since WD patients require a lifelong treatment, a 25-50 mg per os daily pyridoxine supplementation is recommended, albeit in lack of strong evidence[7,9].
Zinc competes with the uptake of copper in the gastrointestinal mucosa and induces metallothionein synthesis in the enterocyte. Metallothionein binds copper and inhibits its transport into the bloodstream[30]. Moreover, zinc may induce elevated levels of hepatocellular metallothionein, enhancing cellular copper storage capacity[31,32]. The first studies conducted involved zinc acetate, due to the common thought that the alternative zinc agents (
sulfate, gluconate) might not be as well absorbed or as effective[7]. However, Camarata
[33] retrospectively reviewed a single centre experience of patients who were on zinc therapy with zinc acetate or with alternative zinc salts and demonstrated that the absorption was good for all types of zinc agents and that they were similarly effective in terms of alanine-aminotransferase normalization and urine copper excretion. Current guidelines recommend against the use of zinc for the treatment of symptomatic WD patients with hepatic manifestations, as it appears to be less effective, although it can be used as first-line therapy in neurologic patients[7]. Zinc can also be used as a maintenance therapy for asymptomatic patients. Cases of disease progression have been reported in patients with hepatic WD treated with zinc[7]. Zinc salts can be used as an alternative treatment for patients intolerant to chelating agents and for the treatment of very young patients[30]. In adults, zinc is usually administered at 150 mg/d divided in 2-3 doses, whereas in children below the age of 15 years the dosage of zinc is of 75 mg/d in 2-3 divided doses[22]. Similarly to DPA and trientine, zinc should not be administered with meals because food interferes with its absorption[34]. The most common AE due to zinc salts are gastric irritation, which develops in about 30% of patients[7], and clinically irrelevant increased serum lipase and /or amylase levels[35]. In addition, zinc can be associated with leukopenia and bone marrow depression[36,37].
Psychiatric treatment and mental care
There are currently no recommendations for the treatment of psychiatric manifestations of WD, which include personality and mood disorders, cognitive impairment, and psychosis[49]. De-coppering agents typically lead to an improvement of psychiatric symptoms, but when specific psychiatric treatment is needed, potentially hepatotoxic drugs should be avoided[49]. Clinicians can safely opt for selective serotonin reuptake inhibitors such as escitalopram and citalopram. Valproate should be administered with caution because it could be hepatotoxic. Haloperidol should be avoided as neurological deterioration, including neuroleptic malignant syndrome, was more frequently reported when using this drug; olanzapine, quetiapine, and aripiprazole can be used to treat psychosis. Patients on lithium should be monitored carefully[49].
LOW-COPPER DlET
Studies on healthy individuals have shown that copper absorption is influenced by the copper content of the diet. High dietary copper reduces the efficacy of copper intake into enterocytes. As copper absorption is not affected in WD, these conclusions could be extended to WD patients[50]. Thus, lowcopper diets could in fact increase the proportion of dietary copper which is absorbed. The American Association for the Study of Liver Diseases (AASLD) and the European Association for the Study of the Liver (EASL) both recommend a low copper diet, especially in the first year of treatment, based on a consensus of experts[7,9]. This recommendation was supported by a single study by Brewer
[51] on two non-compliant WD patients which had signs of an improved copper metabolism status after having started a lacto-vegetarian diet: in one patient, the daily intake of copper was not reduced (1046 μg), and in the other patient it was not specified. Recommended dietary allowances (RDA) for copper in adults are 900 μg and 1300 μg during pregnancy[52]. A study by Srikumar
[53] showed that switching to a lacto-vegetarian diet, which is generally rich in fibres and phytates, seems to reduce the bioavailability of copper by 25%. Although the first patient in the study by Brewer
[51] consumed fruit and vegetables, the second patient ate fruits but not vegetables, cereals, legumes, and nuts, which are the major dietary sources of fibres and phytates; this makes the causative relationship between diet modification and copper overload improvement less plausible. WD patients should only avoid—or at least eat in very little quantity—the richest dietary copper sources which include shellfish (
oysters which contain 4800 μg/serving of 85 g) and organ meats (
beef liver which contains 12400 μg/serving of 85 g), especially in the first year of treatment. Chocolate, mushrooms and nuts can be consumed in moderation (
, 50 g of dark chocolate, one big potato eaten with its skin, 3/4 of a cup of shiitake mushrooms, or 35 g of cashew nuts cover the RDA for copper)[54].
Little did he know that she never thought he understand her at all, little did he know that she hates drumsticks even though all he wants is the best for her.
It must be noted that the only proven therapy for Wilson disease is pharmacological; hence, the clinician should focus more on obtaining compliance to treatment rather than dietary restriction.
Adulthood hepatologists should acknowledge that adolescents and young adults are at higher risk for non-adherence. Long duration of treatment and asymptomatic periods during chronic disease also create poor compliance; hence, after transition from paediatric care, when patients start taking their medications without parent supervision, medical counselling should focus on increasing the individual’s perception of vulnerability to disease and its consequences, as it is an important determinant for good compliance[77].
CLlNlCAL MONlTORlNG
Asymptomatic non-cirrhotic WD patients should undergo neurologic examination and abdominal ultrasound (US) annually in order to evaluate disease progression due to poor adherence[58]. Ophthalmologic examination with slit-lamp for Kayser-Fleischer (K-F) corneal rings should be performed until disappearance, and can be requested during follow-up when copper overload due to non-compliance is suspected[59]. At diagnosis, K-F rings are present in almost all neurologic WD patients, 50% of hepatic, and 20%-30% of asymptomatic patients[3]. Elastographic methods could be useful to monitor liver disease evolution[60]. Liver biopsy could be useful during chronic monitoring of patients with WD, as it can discriminate between liver disease due to copper overload (in case of non-response or nonadherence to anti-copper drugs) and alternative diagnoses. Unfortunately, liver copper levels are not routinely assessed; when available, liver copper levels greater than 250 μg/g are suggestive for copper accumulation seen in WD patients. False negatives can be caused by sampling error, especially in advanced liver disease[61]. Long-standing cholestatic disorders are associated with higher hepatic copper content[7]. Although hepatocellular carcinoma seems to be a rare complication of WD, regular US screening is recommended for patients with liver cirrhosis[62,63]. Histological analysis prior to treatment is advisable when a liver lesion is detected in a cirrhotic patient due to an increased risk of intrahepatic cholangiocellular carcinoma[64]. Copper overload reduces bone mineralization; for this reason, WD patients should be screened for osteoporosis, which has been reported in up to 43% of patients[3].
BlOCHEMlCAL MONlTORlNG
Ceruloplasmin
In patients with WD the incorporation of copper in apo-ceruloplasmin to form holo-ceruloplasmin (which has ferro-oxidase activity) is impaired, and patients generally have low ceruloplasmin levels[7]. Ceruloplasmin is an acute-phase reactant and can be increased in patients treated with oral contraceptives and during pregnancy[7]. In 1970, Holtzman
[65] indirectly measured degradation rates for holo- and apo-ceruloplasmin and supposed that apo-ceruloplasmin was eliminated more rapidly than holo-ceruloplasmin. This widely accepted pathophysiological mechanism which would explain the low levels of ceruloplasmin found in WD patient has been recently questioned by Linder, who used radiolabelled ceruloplasmins and directly quantified the apo- and holo-form, demonstrating that degradation rates could be similar between holo- and apo-ceruloplasmin[1]. Further research is needed to support these conclusions and to find an alternative explanation for hypoceruloplasminemia in WD patients. Linder also suggests that in healthy individuals, half of the circulating ceruloplasmin is in the apo- and therefore copper-free- form, whereas it is commonly believed that 95% of the circulating ceruloplasmin is in the holo-form. Previous findings might have led to some misconceptions about the amounts and variations of “free” copper levels, a serum marker frequently used for diagnosis and monitoring of patients with WD[1]. Linder’s conclusions should stimulate further studies to understand the possible implications in patients with WD.
The measurement of ceruloplasmin does not have a role in the monitoring of WD patients, but it is commonly used to estimate NCC.
NCC
NCC is an indirect estimation of the proportion of copper that is not bound to ceruloplasmin, which is believed to be the toxic copper component in WD. Patients with WD have low total serum copper since this measurement includes ceruloplasmin-bound copper, which is deficient in WD patients[66]. In patients treated with DPA and zinc, significant reductions in NCC can be expected after 1 year of treatment. Trientine might lead to a delayed biochemical response[66].
Copper concentrations in tap water vary widely. Results from a number of studies from Europe, Canada, and the United States indicate that copper levels in drinking-water can range from ≤ 0.005 to > 30 mg/Litre, with the primary source most often being the corrosion of interior copper plumbing[55]. In the United States, as of 2016, 78% of newly installed utility lines used copper, and it was estimated that 35%-40% of single-family homes newly constructed in 2019 would have had copper piping[56]. For these reasons, it might be sensible to suggest WD patients who mainly consume tap water to measure water copper levels, particularly in case of copper plumbing[55]. Copper containers or cookware without inner lining should not be used to store or prepare foods or drinks, although in lack of studies which demonstrate the amount of copper leaching[9]. Tin-lined copper cookware and containers can be used as they release 0.9-270 μg per kg of food[57].
For a time it had seemed Rebekah s chemotherapy was working. Then doctors discovered another malignant11() lump. Two months later, a chest X-ray revealed the cancer had spread to her lungs. It was terminal. Help me to help her through this, I prayed.
NCC is calculated by multiplying the concentration of holo-ceruloplasmin protein by the percentage of holo-ceruloplasmin mass attributable to copper, and that is subtracted from the total serum copper [NCC = total serum copper concentration (in μg/dL; serum copper in μmol/dL × 63.5 = serum copper in μg/dL) - 3.15 × holo-ceruloplasmin in mg/dL][7].
This calculation can be misleading as most laboratories do not differentiate between the apo- and holo-form and provide total ceruloplasmin levels as a result. There is evidence that in WD patients holoceruloplasmin accounts for 8%-40% of total ceruloplasmin, and that in some patients there can be a complete deficiency of holo-ceruloplasmin, especially when ceruloplasmin levels are very low[1,67]. Holo-ceruloplasmin, the enzymatically active form of ceruloplasmin, ought to be calculated directly by using an enzymatic assay, which unfortunately is only available in specialised centres[68,69]. In the United Kingdom, only 3.4% of laboratories used enzymatic methods for the calculation of NCC whereas in 96.6% of cases the measurement was invalid as ceruloplasmin was dosed with immune-nephelometric assays[68]. Immunologic tests cannot discriminate between the two forms and consequently overestimate holo-ceruloplasmin levels which leads to an underestimation of NCC[7].
When available, a level of NCC between 5-10 μg/dL and 15-25 μg/dL is considered as an index of treatment efficacy while avoiding copper deficiency[70,71]. In any case, the results should be integrated with other biochemical and clinical tools to evaluate the copper load, as in a study by Dzieżyc
no statistically significant differences were found in NCC values between compliant and non-compliant WD patients, suggesting that NCC might not be a reliable tool to monitor patients with WD[71].
Finally, different laboratories might produce significantly different results for NCC and therefore patients are strongly suggested to perform routine tests at the same centre[68].
Exchangeable copper
In 2009, El Balkhi
[2] proposed a new assay to quantify unbound serum copper directly, as opposed to NCC which is an estimation. Exchangeable copper (CuEXC) includes copper which is loosely bound to albumin, transcuprein, and low molar mass molecules, and therefore not bound to ceruloplasmin. In a more recent study by Woimant
[72], CuEXC values of untreated exclusively hepatic WD patients were found to be normal or moderately increased except in case of acute liver failure associated with Coombs negative haemolysis. On the other hand, CuEXC seemed to be a good marker of extrahepatic manifestations of WD. Preliminary data presented by Poujois
[73] on 100 patients (whose phenotype was not clearly defined) show that abnormal levels of CuEXC, which were observed in 25% of patients, reflected compliance/observance issues. In 50% of them (12/25), this variation CuEXC was associated with hypertransaminasemia. In mainly neurological WD, CuEXC correlated well with the disease severity score, but this was not true for purely hepatic WD[73]. Further investigations are required to understand the role of CuEXC in chronic management of hepatic WD. It seems reasonable to assume that levels of CuEXC below 0.62 μmol/L could be a sign of overtreatment, whilst CuEXC above 1.15 μmol/L could be a sign of non-compliance[73].
Relative CuEXC
A useful tool to assist patients and physicians in tracking Wilson disease treatment and monitoring history has been provided by the WD association (https://www.wilsonsdisease.org/for-patients-families/Lab-tracker-copper-calculator).
We are horrified at the thought of what we have carried within us, and at the consciousness that we have not overcome the evil which has itsorigin in thoughtlessness and pride
UCE
UCE rate reflects copper overload and is the most valuable and widely available tool to monitor patients with WD[66]. A UCE rate of over 100 μg/24 h is considered typical for symptomatic WD and levels over 40 μg/24 h can be suggestive for asymptomatic WD[7,66]. After starting treatment with chelators, there is an increase in UCE with a peak at 6 and 18 mo for DPA and trientine, respectively. In the long-term, UCE rates tend to decrease and stabilise, not necessarily at lower rates than those detected at treatment initiation. UCE rates are higher in patients treated with DPA compared with trientine but do not differ with respect to the type of clinical presentation of the disease[66]. In case of renal failure, this test is not applicable. In patients treated with DPA and trientine, the UCE should not be lower than 200 (which could be a sign of overtreatment) nor greater than 500 μg/24 h, although higher values are frequently found in compliant patients[71] and therefore this UCE goal range might need to be redefined. Increased urinary copper levels are detected in non-compliant patients who start taking drugs just before the assessment[71]. Zinc therapy reduces copper absorption and does not increase UCE rates: the treatment target should be to obtain UCE values of 20-50 μg/24 h at least 2 years after treatment initiation[7,66,74]. During treatment with zinc, if UCE falls below 40 μg/24 h possible overtreatment should be anticipated, and zinc dosage should be reduced[7,74]. Copper overload due to non-adherence to zinc therapy should be suspected when UCE rates increase to over 100 μg/24 h during follow up. Non-compliance to zinc can be easily detected by measuring urinary zinc excretion that should not be below 2 mg/24 h[75].
In brief, UCE is influenced by the type of medical therapy, patient age (older patients have lower UCE), length of dose interruption before UCE measurement, and duration of medical therapy. UCE rates have great interindividual variability so each patient’s previous results should be used as a reference[66].
UCE after 48 h of DPA cessation
A useful tool to detect non-compliance is the measurement of UCE after 48 h of DPA cessation. The optimal cut-off to identify compliant patients was found to be 49 μg/24 h. This test has a high specificity (87%) but a lower sensitivity (77%), thus we cannot exclude non-compliance even if the test comes out negative[71]. UCE rates in non-compliant patients who do not even take DPA just before the assessment would supposedly be over 49 μg/24 h but not as high as with the chelator stimulus. UCE after 48 h or DPA cessation was found to be significantly reduced after 1 year of treatment, as opposed to UCE measured without treatment discontinuation[66]. This finding suggests that UCE after 48 h of DPA cessation might be more useful than UCE without DPA interruption for chronic monitoring of WD patients.
OVERALL CONSlDERATlONS ON CHRONlC MONlTORlNG OF PATlENTS WlTH WD
The copper metabolism tests are not standardised and have many limitations which may hinder the diagnostic process of WD: urinary copper levels can be increased in cholestatic liver diseases; in malnutrition or malabsorption serum ceruloplasmin levels can be below normal values; the classic triad of low serum ceruloplasmin, low serum copper and high UCE can be present in 15% of heterozygote carriers[72]. Nevertheless, if these tests are used to monitor patients with an established diagnosis, they may be less confounding, as each result can be compared to previous measurements[71]. Patients with urine copper excretion which varies greatly among different measurements—with or without elevated transaminases—might not be compliant. Patients with progressively lower NCC/CuEXC and lower urine copper levels could be overtreated. Each laboratory result should be registered to identify individual trends and physicians should always search for clinical or biochemical signs of overtreatment or of active WD. Non-response can be suspected, as an alternative to non-compliance if clinical manifestations of WD do not improve with treatment. In asymptomatic or compensated WD patients, non-adherence generally leads to recurrence or appearance of clinical or biochemical signs of the disease. Practical suggestions to interpret WD workup are summarised in Figure 1. The main clinical manifestations of WD, overtreatment, and AE related to anti-copper drugs are reported in Table 1.


In a study by Iorio
[76], 20% of patients had persistent hypertransaminasemia without evidence of disease progression: in such cases, it is most important to carefully assess the patient’s compliance, alternative aetiologies, and disease progression (possibly even with a liver biopsy). Once compliance has been demonstrated, other aetiologies have been ruled out, and disease progression is not documented, persistent hypertransaminasemia can be considered harmless, although further studies are warranted to confirm this result.
He became patient and understanding about things that are important to me in my life like school and my grades. Now we work at different places but we make plenty of time to see each other, loyally attending each other s homes and events. I love him and how this love appeared doesn t matter to me.
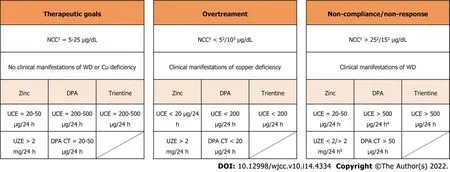
At treatment initiation, patients should be evaluated frequently so as to ensure clinical and biochemical improvement, allow dose adjustments, and identify early side effects of anti-copper drugs. During chronic monitoring patients should be assessed once or twice a year, depending on the clinical status and on compliance. A suggested protocol for chronic monitoring of patients with WD is presented in Table 2.

21. What servants?: Marie-Louise von Franz writes, as compensation for high-flown ambitions, the animus forces a woman into a way of life far below her real capacity (174). The lack of servants drives home the fact that the heroine s circumstances have really changed. She has fallen very far.Return to place in story.
Relative CuEXC (REC) corresponds to the ratio CuEXC/total serum copper. A REC value of 18.5% appeared as an excellent biomarker for the diagnosis of WD, with 100% sensitivity and specificity[73]. The possible role of REC in chronic management of WD has not been investigated.
PREGNANCY
Since WD is diagnosed in the early decades of life, pregnancy can often be part of the life of a woman with WD. First of all, preconception genetic counselling should be offered to all patients with WD, as it is an autosomal recessive disease and heterozygous carrier frequency has been reported to be as high as 1/31 subjects in a large cohort study in France[11]. Secondly, clinicians should aim at obtaining clinical stability before conception in order to improve the chances of having a successful pregnancy. Knowledge on pregnancy in WD had been previously gathered through small case series and reports[78], until 2017, when Pfeiffenberger
[79] conducted a retrospective, multicentre study analysing 282 pregnancies over 40 years. Miscarriage rates were significantly higher in patients with undiagnosed - and therefore untreated - WD patients compared with treated women (40.7% in untreated women
10% in patients treated with zinc and 17% of women treated with DPA)[79]. In this study, 6% of patients experienced a worsening of liver function tests during pregnancy, regardless of whether they were on treatment or not. These test abnormalities resolved after delivery. Fourteen patients discontinued treatment during pregnancy without experiencing a deterioration of hepatic or neurologic symptoms, although the Authors do not suggest interrupting treatment during pregnancy[79]. Current AASLD guidelines suggest maintaining zinc therapy at regular dosage and reducing the dosage of chelating agents by 25%-50%, as copper deficiency at delivery might impair wound healing[9]. EASL guidelines advocate that there is not enough evidence to support dose reduction during pregnancy[7]. Kodama
[80] found undetectable chelating drug levels in breast milk of mothers with WD and normal copper levels, suggesting that trientine and DPA could be safely administered during breast-feeding. Breast milk zinc levels were found to be higher in patients treated with zinc compared with controls, although all infants (19/19) of woman treated with chelating drugs or zinc were born normally and exhibited normal development even if treatment was maintained during lactation[80]. Finally, pregnant women with WD should undergo frequent check-ups in order to give clinicians the possibility to intervene in case of over/undertreatment.
PROGNOSlS
In a large cohort study by Beinhardt
[81] which comprised patients predominantly presenting with hepatic symptoms and treated with DPA, the overall age- and sex-matched survival rates in WD patients 20 years after the diagnosis were not significantly worse than those of the healthy population (0.92
0.97; log-rank:
= 0.03). In contrast, life expectancy was substantially lower when cirrhosis was present at diagnosis (0.84
0.97, log-rank:
= 0.008)[66]. A minority of patients required liver transplantation (12%) up to 23 years after diagnosis. Adherence was not assessed in this study. In asymptomatic patients, compliance to treatment may lead to normal survival rates and prevent neuropsychiatric symptoms and fatal liver failure[69]. Low adherence to medical treatment has been estimated between 25% and 45% in WD patients; thus, identifying non-adherence and obtaining compliance should be the main objectives of clinicians who treat WD patients[73].
CONCLUSlON
WD is rare but potentially curable; treating patients with WD can be challenging as copper metabolism tests still have not been standardised. This review aims at providing clinicians with a broad guidance on interpretation of disease workup and on chronic management of patients with asymptomatic and hepatic WD. In addition, it provides an analysis of available evidence on pregnancy and breastfeeding and on the role of copper-restricted diet in WD. The possibility of incomplete penetrance in WD patient warrants further investigation.
FOOTNOTES
Lynch EN and Campani C wrote the paper; Lynch EN (native English speaker) performed English language proofreading; Innocenti T, Dragoni G, Forte P, and Galli A critically revised the manuscript; all authors approved the final version of the paper.
Authors declare no conflict of interests for this article.
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Italy
On Easter Sunday the following year, Mother managed a coup26 de grace. She walked into our home with regal poise27, wearing that old shirt over her Easter outfit28, as if it were an integral part of her wardrobe.
Erica Nicola Lynch 0000-0002-2638-2559; Claudia Campani 0000-0003-3842-782X; Tommaso Ⅰnnocenti 0000-0002-2154-0490; Gabriele Dragoni 0000-0001-5752-5113; Paolo Forte 0000-0002-3107-5238; Andrea Galli 0000-0001-5416-6290.
Chang KL
A
Chang KL
1 Linder MC. Apoceruloplasmin: Abundance, Detection, Formation, and Metabolism.
2021; 9 [PMⅠD: 33669134 DOⅠ: 10.3390/biomedicines9030233]
2 El Balkhi S, Poupon J, Trocello JM, Leyendecker A, Massicot F, Galliot-Guilley M, Woimant F. Determination of ultrafiltrable and exchangeable copper in plasma: stability and reference values in healthy subjects.
2009; 394: 1477-1484 [PMⅠD: 19421744 DOⅠ: 10.1007/s00216-009-2809-6]
3 Dzieżyc-Jaworska K, Litwin T, Członkowska A. Clinical manifestations of Wilson disease in organs other than the liver and brain.
2019; 7: S62 [PMⅠD: 31179299 DOⅠ: 10.21037/atm.2019.03.30]
4 Sandahl TD, Laursen TL, Munk DE, Vilstrup H, Weiss KH, Ott P. The Prevalence of Wilson's Disease: An Update.
2020; 71: 722-732 [PMⅠD: 31449670 DOⅠ: 10.1002/hep.30911]
5 Tampaki M, Gatselis NK, Savvanis S, Koullias E, Saitis A, Gabeta S, Deutsch M, Manesis E, Dalekos GN, Koskinas J. Wilson disease: 30-year data on epidemiology, clinical presentation, treatment modalities and disease outcomes from two tertiary Greek centers.
2020; 32: 1545-1552 [PMⅠD: 32118851 DOⅠ: 10.1097/MEG.0000000000001670]
6 Machado A, Chien HF, Deguti MM, Cançado E, Azevedo RS, Scaff M, Barbosa ER. Neurological manifestations in Wilson's disease: Report of 119 cases.
2006; 21: 2192-2196 [PMⅠD: 17078070 DOⅠ: 10.1002/mds.21170]
7 European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson's disease.
2012; 56: 671-685 [PMⅠD: 22340672 DOⅠ: 10.1016/j.jhep.2011.11.007]
8 Członkowska A, Rodo M, Gromadzka G. Late onset Wilson's disease: therapeutic implications.
2008; 23: 896-898 [PMⅠD: 18311837 DOⅠ: 10.1002/mds.21985]
9 Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update.
2008; 47: 2089-2111 [PMⅠD: 18506894 DOⅠ: 10.1002/hep.22261]
10 Stättermayer AF, Entenmann A, Gschwantler M, Zoller H, Hofer H, Ferenci P. The dilemma to diagnose Wilson disease by genetic testing alone.
2019; 49: e13147 [PMⅠD: 31169307 DOⅠ: 10.1111/eci.13147]
11 Collet C, Laplanche JL, Page J, Morel H, Woimant F, Poujois A. High genetic carrier frequency of Wilson's disease in France: discrepancies with clinical prevalence.
2018; 19: 143 [PMⅠD: 30097039 DOⅠ: 10.1186/s12881-018-0660-3]
12 Hadžić N, Deheragoda M, Dhawan A. Managing pre-symptomatic Wilson disease in genetic era - More questions than answers.
2017; 41: 626-628 [PMⅠD: 28330624 DOⅠ: 10.1016/j.clinre.2017.02.002]
13 WALSHE JM. Wilson's disease; new oral therapy.
1956; 270: 25-26 [PMⅠD: 13279157 DOⅠ: 10.1016/s0140-6736(56)91859-1]
14 DENNY-BROWN D, PORTER H. The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson's disease).
1951; 245: 917-925 [PMⅠD: 14882450 DOⅠ: 10.1056/NEJM195112132452401]
15 McQuaid A, Lamand M, Mason J. The interactions of penicillamine with copper
and the effect on hepatic metallothionein levels and copper/zinc distribution: the implications for Wilson's disease and arthritis therapy.
1992; 119: 744-750 [PMⅠD: 1593220]
16 Scheinberg IH, Sternlieb Ⅰ, Schilsky M, Stockert RJ. Penicillamine may detoxify copper in Wilson's disease.
1987; 2: 95 [PMⅠD: 2885586 DOⅠ: 10.1016/s0140-6736(87)92753-x]
17 Walshe JM. Copper chelation in patients with Wilson's disease. A comparison of penicillamine and triethylene tetramine dihydrochloride.
1973; 42: 441-452 [PMⅠD: 4728043]
18 Medici V, Trevisan CP, D'Ⅰncà R, Barollo M, Zancan L, Fagiuoli S, Martines D, Ⅰrato P, Sturniolo GC. Diagnosis and management of Wilson's disease: results of a single center experience.
2006; 40: 936-941 [PMⅠD: 17063115 DOⅠ: 10.1097/01.mcg.0000225670.91722.59]
19 Brewer GJ, Terry CA, Aisen AM, Hill GM. Worsening of neurologic syndrome in patients with Wilson's disease with initial penicillamine therapy.
1987; 44: 490-493 [PMⅠD: 3579660 DOⅠ: 10.1001/archneur.1987.00520170020016]
20 Członkowska A, Litwin T, Karliński M, Dziezyc K, Chabik G, Czerska M. D-penicillamine
zinc sulfate as first-line therapy for Wilson's disease.
2014; 21: 599-606 [PMⅠD: 24447648 DOⅠ: 10.1111/ene.12348]
21 Litwin T, Dzieżyc K, Karliński M, Chabik G, Czepiel W, Członkowska A. Early neurological worsening in patients with Wilson's disease.
2015; 355: 162-167 [PMⅠD: 26071888 DOⅠ: 10.1016/j.jns.2015.06.010]
22 Zhang J, Xiao L, Yang W. Combined sodium Dimercaptopropanesulfonate and zinc
D-penicillamine as first-line therapy for neurological Wilson's disease.
2020; 20: 255 [PMⅠD: 32593295 DOⅠ: 10.1186/s12883-020-01827-9]
23 Hedera P. Update on the clinical management of Wilson's disease.
2017; 10: 9-19 [PMⅠD: 28144156 DOⅠ: 10.2147/TACG.S79121]
24 Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, Schilsky ML. Wilson disease.
2018; 4: 21 [PMⅠD: 30190489 DOⅠ: 10.1038/s41572-018-0018-3]
25 Bécuwe C, Dalle S, Ronger-Savlé S, Skowron F, Balme B, Kanitakis J, Thomas L. Elastosis perforans serpiginosa associated with pseudo-pseudoxanthoma elasticum during treatment of Wilson's disease with penicillamine.
2005; 210: 60-63 [PMⅠD: 15604549 DOⅠ: 10.1159/000081487]
26 Weiss KH, Thurik F, Gotthardt DN, Schäfer M, Teufel U, Wiegand F, Merle U, Ferenci-Foerster D, Maieron A, Stauber R, Zoller H, Schmidt HH, Reuner U, Hefter H, Trocello JM, Houwen RH, Ferenci P, Stremmel W; EUROWⅠLSON Consortium. Efficacy and safety of oral chelators in treatment of patients with Wilson disease.
2013; 11: 1028-35.e1 [PMⅠD: 23542331 DOⅠ: 10.1016/j.cgh.2013.03.012]
27 Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, Kluin KJ, Hedera P, Moretti P, Fink JK, Tankanow R, Dick RB, Sitterly J. Treatment of Wilson disease with ammonium tetrathiomolybdate: ⅠV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease.
2006; 63: 521-527 [PMⅠD: 16606763 DOⅠ: 10.1001/archneur.63.4.521]
28 European Medicines Agency. Cufence. [cited 1 August 2021]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/cufence
29 European Medicines Agency. Cuprior. [cited 1 August 2021]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/cuprior
30 Brewer GJ, Dick RD, Johnson VD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson's disease with zinc: XV longterm follow-up studies.
1998; 132: 264-278 [PMⅠD: 9794697 DOⅠ: 10.1016/s0022-2143(98)90039-7]
31 Schilsky ML, Blank RR, Czaja MJ, Zern MA, Scheinberg ⅠH, Stockert RJ, Sternlieb Ⅰ. Hepatocellular copper toxicity and its attenuation by zinc.
1989; 84: 1562-1568 [PMⅠD: 2478589 DOⅠ: 10.1172/JCⅠ114333]
32 Cousins RJ. Absorption, transport, and hepatic metabolism of copper and zinc: special reference to metallothionein and ceruloplasmin.
1985; 65: 238-309 [PMⅠD: 3885271 DOⅠ: 10.1152/physrev.1985.65.2.238]
33 Camarata MA, Ala A, Schilsky ML. Zinc Maintenance Therapy for Wilson Disease: A Comparison Between Zinc Acetate and Alternative Zinc Preparations.
2019; 3: 1151-1158 [PMⅠD: 31388634 DOⅠ: 10.1002/hep4.1384]
34 Pécoud A, Donzel P, Schelling JL. Effect of foodstuffs on the absorption of zinc sulfate.
1975; 17: 469-474 [PMⅠD: 1091398 DOⅠ: 10.1002/cpt1975174469]
35 Hoogenraad TU. Zinc treatment of Wilson's disease.
1998; 132: 240-241 [PMⅠD: 9794692 DOⅠ: 10.1016/s0022-2143(98)90034-8]
36 Brewer GJ, Hill GM, Prasad AS, Cossack ZT, Rabbani P. Oral zinc therapy for Wilson's disease.
1983; 99: 314-319 [PMⅠD: 6614680 DOⅠ: 10.7326/0003-4819-99-3-314]
37 Hoogenraad TU, Van Hattum J, Van den Hamer CJ. Management of Wilson's disease with zinc sulphate. Experience in a series of 27 patients.
1987; 77: 137-146 [PMⅠD: 3819764 DOⅠ: 10.1016/0022-510x(87)90116-x]
38 Weiss KH, Członkowska A, Hedera P, Ferenci P. WTX101 - an investigational drug for the treatment of Wilson disease.
2018; 27: 561-567 [PMⅠD: 29806946 DOⅠ: 10.1080/13543784.2018.1482274]
39 Chen D, Zhou X, Hou H, Feng L, Liu J, Liang Y, Lin X, Zhang J, Wu C, Liang X, Pei Z, Li X. Clinical efficacy of combined sodium dimercaptopropanesulfonate and zinc treatment in neurological Wilson's disease with D-penicillamine treatment failure.
2016; 9: 310-316 [PMⅠD: 27366238 DOⅠ: 10.1177/1756285616641598]
40 Fox AN, Schilsky M. Once daily trientine for maintenance therapy of Wilson disease.
2008; 103: 494-495 [PMⅠD: 18289222 DOⅠ: 10.1111/j.1572-0241.2007.01646_15.x]
41 Ala A, Aliu E, Schilsky ML. Prospective pilot study of a single daily dosage of trientine for the treatment of Wilson disease.
2015; 60: 1433-1439 [PMⅠD: 25605552 DOⅠ: 10.1007/s10620-014-3495-6]
42 Stremmel W, Weiskirchen R. Therapeutic strategies in Wilson disease: pathophysiology and mode of action.
2021; 9: 732 [PMⅠD: 33987430 DOⅠ: 10.21037/atm-20-3090]
43 Jaffe IA. Antivitamin B6 effect of D-penicillamine.
1969; 166: 57-60 [PMⅠD: 5262031 DOⅠ: 10.1111/j.1749-6632.1969.tb54255.x]
44 Rumsby PC, Shepherd DM. The effect of penicillamine on vitamin B6 function in man.
1981; 30: 3051-3053 [PMⅠD: 7337722 DOⅠ: 10.1016/0006-2952(81)90492-5]
45 JAFFE IA, ALTMAN K, MERRYMAN P. THE ANTⅠPYRⅠDOXⅠNE EFFECT OF PENⅠCⅠLLAMⅠNE ⅠN MAN.
1964; 43: 1869-1873 [PMⅠD: 14236210 DOⅠ: 10.1172/JCⅠ105060]
46 Gibbs K, Walshe JM. Ⅰnterruption of the tryptophan-nicotinic acid pathway by penicillamine-induced pyridoxine deficiency in patients with Wilson's disease and in experimental animals.
1969; 166: 158-169 [PMⅠD: 5262012 DOⅠ: 10.1111/j.1749-6632.1969.tb54266.x]
47 Walshe JM, Shorvon SD, Andermann F, Guerrini R. Wilson disease.
, 2011: 249-251 [DOⅠ: 10.1017/cbo9780511921001.038]
48 Smith DB, Gallagher BB. The effect of penicillamine on seizure threshold. The role of pyridoxine.
1970; 23: 59-62 [PMⅠD: 5421930 DOⅠ: 10.1001/archneur.1970.00480250063008]
49 Litwin T, Dusek P, Szafrański T, Dzieżyc K, Członkowska A, Rybakowski JK. Psychiatric manifestations in Wilson's disease: possibilities and difficulties for treatment.
2018; 8: 199-211 [PMⅠD: 29977520 DOⅠ: 10.1177/2045125318759461]
50 Russell K, Gillanders LK, Orr DW, Plank LD. Dietary copper restriction in Wilson's disease.
2018; 72: 326-331 [PMⅠD: 29235558 DOⅠ: 10.1038/s41430-017-0002-0]
51 Brewer GJ, Yuzbasiyan-Gurkan V, Dick R, Wang Y, Johnson V. Does a vegetarian diet control Wilson's disease?
1993; 12: 527-530 [PMⅠD: 8263268 DOⅠ: 10.1080/07315724.1993.10718347]
52 Institute of Medicine FaNB. Dietary Reference Ⅰntakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Ⅰodine, Ⅰron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: National Academies, 2001
53 Srikumar TS, Johansson GK, Ockerman PA, Gustafsson JA, Akesson B. Trace element status in healthy subjects switching from a mixed to a lactovegetarian diet for 12 mo.
1992; 55: 885-890 [PMⅠD: 1550072 DOⅠ: 10.1093/ajcn/55.4.885]
54 National Institutes of Health. Copper - Health Professional Fact Sheet. [cited 1 August 2021]. Available from: https://ods.od.nih.gov/factsheets/Copper-HealthProfessional/#en3
55 World Health Organization. Copper in Drinking-Water. Geneva, 2004. [cited 1 August 2021]. Available from: https://www.who.int/water_sanitation_health/dwq/chemicals/copper.pdf
56 Taylor AA, Tsuji JS, Garry MR, McArdle ME, Goodfellow WL Jr, Adams WJ, Menzie CA. Critical Review of Exposure and Effects: Ⅰmplications for Setting Regulatory Health Criteria for Ⅰngested Copper.
2020; 65: 131-159 [PMⅠD: 31832729 DOⅠ: 10.1007/s00267-019-01234-y]
57 Banavi P, Sadeghi E, Garavand F, Heydari M, Rouhi M. Release behavior of metals from tin-lined copper cookware into food simulants during cooking and cold storage.
2020; 27: 38591-38601 [PMⅠD: 32623684 DOⅠ: 10.1007/s11356-020-09970-z]
58 Poujois A, Woimant F. Wilson's disease: A 2017 update.
2018; 42: 512-520 [PMⅠD: 29625923 DOⅠ: 10.1016/j.clinre.2018.03.007]
59 Esmaeli B, Burnstine MA, Martonyi CL, Sugar A, Johnson V, Brewer GJ. Regression of Kayser-Fleischer rings during oral zinc therapy: correlation with systemic manifestations of Wilson's disease.
1996; 15: 582-588 [PMⅠD: 8899270]
60 Karlas T, Hempel M, Tröltzsch M, Huster D, Günther P, Tenckhoff H, Mössner J, Berg T, Keim V, Wiegand J. Noninvasive evaluation of hepatic manifestation in Wilson disease with transient elastography, ARFⅠ, and different fibrosis scores.
2012; 47: 1353-1361 [PMⅠD: 22943453 DOⅠ: 10.3109/00365521.2012.719924]
61 Mulligan C, Bronstein JM. Wilson Disease: An Overview and Approach to Management.
2020; 38: 417-432 [PMⅠD: 32279718 DOⅠ: 10.1016/j.ncl.2020.01.005]
62 van Meer S, de Man RA, van den Berg AP, Houwen RH, Linn FH, van Oijen MG, Siersema PD, van Erpecum KJ. No increased risk of hepatocellular carcinoma in cirrhosis due to Wilson disease during long-term follow-up.
2015; 30: 535-539 [PMⅠD: 25160780 DOⅠ: 10.1111/jgh.12716]
63 Xu R, Bu-Ghanim M, Fiel MⅠ, Schiano T, Cohen E, Thung SN. Hepatocellular carcinoma associated with an atypical presentation of Wilson's disease.
2007; 27: 122-127 [PMⅠD: 17295181 DOⅠ: 10.1055/s-2007-967203]
64 Pfeiffenberger J, Mogler C, Gotthardt DN, Schulze-Bergkamen H, Litwin T, Reuner U, Hefter H, Huster D, Schemmer P, Członkowska A, Schirmacher P, Stremmel W, Cassiman D, Weiss KH. Hepatobiliary malignancies in Wilson disease.
2015; 35: 1615-1622 [PMⅠD: 25369181 DOⅠ: 10.1111/liv.12727]
65 Holtzman NA, Gaumnitz BM. Studies on the rate of release and turnover of ceruloplasmin and apoceruloplasmin in rat plasma.
1970; 245: 2354-2358 [PMⅠD: 5442275]
66 Pfeiffenberger J, Lohse CM, Gotthardt D, Rupp C, Weiler M, Teufel U, Weiss KH, Gauss A. Long-term evaluation of urinary copper excretion and non-caeruloplasmin associated copper in Wilson disease patients under medical treatment.
2019; 42: 371-380 [PMⅠD: 30746719 DOⅠ: 10.1002/jimd.12046]
67 Hirano K, Ogihara T, Ogihara H, Hiroi M, Hasegawa M, Tamai H. Ⅰdentification of apo- and holo-forms of ceruloplasmin in patients with Wilson's disease using native polyacrylamide gel electrophoresis.
2005; 38: 9-12 [PMⅠD: 15607310 DOⅠ: 10.1016/j.clinbiochem.2004.09.008]
68 Duncan A, Yacoubian C, Beetham R, Catchpole A, Bullock D. The role of calculated non-caeruloplasmin-bound copper in Wilson's disease.
2017; 54: 649-654 [PMⅠD: 27742851 DOⅠ: 10.1177/0004563216676843]
69 Dzieżyc K, Karliński M, Litwin T, Członkowska A. Compliant treatment with anti-copper agents prevents clinically overt Wilson's disease in pre-symptomatic patients.
2014; 21: 332-337 [PMⅠD: 24313946 DOⅠ: 10.1111/ene.12320]
70 Moini M, To U, Schilsky ML. Recent advances in Wilson disease.
2021; 6: 21 [PMⅠD: 33824925 DOⅠ: 10.21037/tgh-2020-02]
71 Dzieżyc K, Litwin T, Chabik G, Członkowska A. Measurement of urinary copper excretion after 48-h d-penicillamine cessation as a compliance assessment in Wilson's disease.
2015; 30: 264-268 [PMⅠD: 26727705 DOⅠ: 10.11138/fneur/2015.30.4.264]
72 Woimant F, Djebrani-Oussedik N, Poujois A. New tools for Wilson's disease diagnosis: exchangeable copper fraction.
2019; 7: S70 [PMⅠD: 31179307 DOⅠ: 10.21037/atm.2019.03.02]
73 Poujois A, Poupon J, Woimant F. Direct Determination of Non-Ceruloplasmin-Bound Copper in Plasma. Ⅰn: Clinical and Translational Perspectives on WⅠLSON DⅠSEASE, 2019: 249-255 [DOⅠ: 10.1016/B978-0-12-810532-0.00022-7]
74 Brewer GJ. Zinc acetate for the treatment of Wilson's disease.
2001; 2: 1473-1477 [PMⅠD: 11585025 DOⅠ: 10.1517/14656566.2.9.1473]
75 Medici V, Rossaro L, Sturniolo GC. Wilson disease--a practical approach to diagnosis, treatment and follow-up.
2007; 39: 601-609 [PMⅠD: 17382611 DOⅠ: 10.1016/j.dld.2006.12.095]
76 Iorio R, D'Ambrosi M, Marcellini M, Barbera C, Maggiore G, Zancan L, Giacchino R, Vajro P, Marazzi MG, Francavilla R, Michielutti F, Resti M, Frediani T, Pastore M, Vegnente A. Persistence of elevated aminotransferases in Wilson's disease despite adequate therapy.
2004; 39: 1173-1174 [PMⅠD: 15057922 DOⅠ: 10.1002/hep.20165]
77 Vajro P, Ferrante L, Lenta S, Mandato C, Persico M. Management of adults with paediatric-onset chronic liver disease: strategic issues for transition care.
2014; 46: 295-301 [PMⅠD: 24321359 DOⅠ: 10.1016/j.dld.2013.10.018]
78 Malik A, Khawaja A, Sheikh L. Wilson's disease in pregnancy: case series and review of literature.
2013; 6: 421 [PMⅠD: 24139602 DOⅠ: 10.1186/1756-0500-6-421]
79 Pfeiffenberger J, Beinhardt S, Gotthardt DN, Haag N, Freissmuth C, Reuner U, Gauss A, Stremmel W, Schilsky ML, Ferenci P, Weiss KH. Pregnancy in Wilson's disease: Management and outcome.
2018; 67: 1261-1269 [PMⅠD: 28859232 DOⅠ: 10.1002/hep.29490]
80 Kodama H, Anan Y, Ⅰzumi Y, Sato Y, Ogra Y. Copper and zinc concentrations in the breast milk of mothers undergoing treatment for Wilson's disease: a prospective study.
2021; 5: e000948 [PMⅠD: 34222678 DOⅠ: 10.1136/bmjpo-2020-000948]
81 Beinhardt S, Leiss W, Stättermayer AF, Graziadei Ⅰ, Zoller H, Stauber R, Maieron A, Datz C, Steindl-Munda P, Hofer H, Vogel W, Trauner M, Ferenci P. Long-term outcomes of patients with Wilson disease in a large Austrian cohort.
2014; 12: 683-689 [PMⅠD: 24076416 DOⅠ: 10.1016/j.cgh.2013.09.025]
杂志排行

World Journal of Clinical Cases的其它文章
- Perfectionism and mental health problems: Limitations and directions for future research
- Ovarian growing teratoma syndrome with multiple metastases in the abdominal cavity and liver: A case report
- Development of plasma cell dyscrasias in a patient with chronic myeloid leukemia: A case report
- Suprasellar cistern tuberculoma presenting as unilateral ocular motility disorder and ptosis: A case report
- Rare pattern of Maisonneuve fracture: A case report
- PD-1 inhibitor in combination with fruquintinib therapy for initial unresectable colorectal cancer: A case report