基于部分DNA甲基化预测肿瘤浸润免疫细胞的比例
2022-05-31宋春晖秦玉芳陈明
宋春晖,秦玉芳,陈明
1.上海海洋大学信息学院,上海 201306;2.农业农村部渔业信息重点实验室,上海 201306
前言
DNA 甲基化状态是基因组研究中一个突出的表观遗传学标记。随着全基因组DNA甲基化数据的获得,大量研究为DNA 甲基化在细胞过程和疾病如肾癌[1]、结直肠癌[2]中发挥的作用提供了证据,DNA 甲基化状态为疾病的诊断和治疗提供了很好的帮助。
肿瘤微环境中存在的浸润性免疫细胞在癌症进展、患者存活和对症治疗中起重要作用[3]。组织混合物细胞成分测定的实验方法有流式细胞术和尖端单细胞技术,如Drop-seq[4]、10X 基因组学和sci-RNAseq[5],但是实验价格昂贵、劳动强度大,需要新鲜的组织,并且对细胞分离过程中的技术变化敏感。因此除了实验方法,采用计算方法预测肿瘤组织中细胞成分的含量十分必要。
基于转录组的基因表达数据对复杂混合物(包括实体瘤)进行分析进而推断细胞类型比例的方法目前有了一些研究[6-9]。Newman 等[6]采用支持向量回归的方法CIBERSORT,利用微阵列数据对未知的复杂混合物进行细胞类型比例估计。基于细胞类型特异性mRNA 含量的重组,EPIC 考虑特征差异显著的细胞类型,从大量的肿瘤转录组表达数据中估计肿瘤和免疫细胞类型的比例[7]。然而由于临床上组织样品的化学固定[10],通过转录组分析方法测量的RNA 分子更容易降解,而DNA 甲基化是更稳定的分子且具有高度的细胞类型特异性[11-12],因此基于DNA 甲基化的方法成为细胞反卷积的一种更有效的替代方法。目前,对混合物中细胞含量的估计主要分为两类,一类是基于参考的方法,Houseman等[13]将混合物样本作为组成细胞类型的DNA甲基化与比例的加权组合[14],利用约束投影/二次规划推断混合物样本中的细胞类型比例;Teschendorff 等[15]利用来自NIH 表观基因组的细胞类型特异性高灵敏位点信息构建甲基化参考数据来推断全血样本中细胞类型的比例。另一类是基于无参考的方法,最初的两种方法FaST-LMM-EWASher 和RefFreeEWAS 分别由James 等[16]和Houseman 等[17]提出,随后Pavlo 等[18]在此基础上,基于约束性非负矩阵分解并结合一种新的生物相关正则化函数开发了MeDeCom,并用于预测混合物中细胞类型的比例;Rahmani等[19]基于主成分分析开发了ReFACTor,该方法不需要细胞计数的先验知识,提供了细胞类型组成的改进估计。虽然原则上基于无参考的方法可以应用于任何组织,但这种方法的预测准确率较低;基于参考的方法能获得较高的细胞比例预测准确率,然而在实际临床上往往只知道一部分细胞类型的甲基化,即部分参考的情况,因此可以利用容易获得的肿瘤混合物中部分细胞类型的表观基因组信息去推断出所有组成细胞类型的比例。基于部分参考的方法已成功应用于基于转录组数据的肿瘤浸润免疫细胞的分解[20]。
本研究基于肿瘤组织中已知细胞类型的甲基化,利用非负矩阵分解的框架去估计所有细胞类型的比例,简记为MethyPR。对模拟数据的评估表明本研究的方法较现有方法在识别细胞类型比例上有了明显的提高;其次,在体外制备的混合物上验证了本研究的方法能很好地还原出所有细胞类型的比例;最后,将MethyPR 应用于癌症基因组图谱(TCGA)甲基化数据,能很好地识别出癌症特异性肿瘤浸润性免疫细胞,为靶向免疫治疗的设计提供依据。
1 材料与方法
1.1 数据集获取
1.1.1 模拟混合物数据本研究从基因表达综合数据库(GEO)下载了6 种纯化免疫细胞[CD4+T 细胞、CD8+T细胞、自然杀伤细胞、B细胞、单核细胞(Mon)和粒细胞(Gra)(GSE35069)]和一种乳腺癌细胞(MCF-7)(GSE44837)的甲基化谱,并按照一定比例产生免疫细胞和肿瘤细胞的混合物。
1.1.2 实验获得的真实样本数据真实样本的甲基化数据以及组成样本的细胞类型甲基化数据来源于Onuchic等[21]的研究,具体来说,该数据集包含两个样本集和构成样本的细胞类型甲基化数据。第一个样本集由6个样本组成,分别由3个成对细胞系的组合组成(MCF-7/HMEC、MCF-7/CD8+T 细胞和MCF-7/CAF),分别按75%:25%和95%:5%的比例混合组成。第二个样本集是由亚硫酸氢盐测序生成的29个乳腺肿瘤样本组成,每个样本由不同的乳腺癌细胞系、一种正常乳腺细胞系(HMEC)、一种成纤维细胞系(CAF)和一种免疫细胞(CD8+T)组成,同时使用H&E染色,估计每个样本的癌性、正常、成纤维和免疫细胞的比例。该数据集还包含了构成样本的细胞系的甲基化数据,分别是6种不同的乳腺癌细胞系(MCF-7、T47D、MDA-MB-231、MDA-MB-361、HCC1954、HCC1569)、HMEC、CAF和CD8+T细胞。
1.1.3 TCGA癌症样本数据本研究使用GDC客户端工具从GDC 数据门户(https://gdc.nci.nih.gov)下载胸腺瘤、结直肠癌、急性髓系肿瘤和弥漫性大B 细胞淋巴瘤样本的Infinium HumanMethylation 450 芯片的三级甲基化数据。另外,从Arneson 等[22]的研究得到免疫细胞的甲基化数据,该数据集包含11 种细胞类型(单核细胞、树突状细胞、巨噬细胞、中性粒细胞、嗜酸性粒细胞、调控性T 细胞、幼稚T 细胞、记忆T 细胞、CD8+T 细胞、自然杀伤细胞和B 细胞)的甲基化数据(GSE35069、GSE59250、GSE71837),并且按照文献[22]的方法,采用单核细胞作为桥接细胞类型纠正批次效应,最终得到419个甲基化位点处的肿瘤浸润性免疫细胞甲基化数据。
1.2 数据预处理
对于下载得到的高维甲基化数据,首先排除甲基化位点缺失超过10%的样本以及样本缺失超过10%的甲基化位点。然后,用R 包“ChAMP”包进行一系列处理:使用ChAMP.impute 方法对缺失值进行填充,使用ChAMP.filter 方法过滤常染色体上与性相关的和位于X、Y 染色体上的位点,以避免任何与性别相关的信号,同时过滤所有与单核苷酸多态性(Single Nucleotide Polymorphism, SNP)重叠的和报道为交叉反应的位点[23]。例如,对于模拟实验中6种免疫细胞的高维甲基化数据,排除6个在每种细胞中缺失值超过10%的位点,接下来过滤10 028个与性相关和位于X、Y 染色体上的位点,使用一般的450K SNP列表过滤了59 901个SNP重叠的位点,并移除了11个交叉反应的位点,最终得到6种免疫细胞对应的412 481个位点处的甲基化水平。
为了获得对每种细胞类型差异显著的位点,对上述数据进行进一步处理,使用ChAMP.DMP方法提取细胞类型中差异显著的位点[24]。接下来,遵循文献[25]对上述得到的数据集中的每种细胞类型的甲基化水平与其他细胞类型甲基化水平进行t 检验比较,得到差异显著(P<0.000 1)的甲基化位点,按照P值进行降序排列,选择出1 000个甲基化位点,并选择最终的甲基化位点为每次t检验中选择出的位点的交集。例如:对于模拟实验中的6 种免疫细胞,通过“ChAMP”步骤的处理,得到25 820 个对于每种细胞类型差异显著的位点,经过t检验的处理后,最终得到包含412个显著差异的位点处的甲基化水平。
1.3 模型
1.3.1 模型建立MethyPR 的模型如图1 所示。M∈Rm×n是来自k个细胞类型组成的一个行为m个位点,列对应n个样本的DNA 甲基化水平矩阵。其中Mij∈M,表示第i个位点在第j个样本的甲基化探针占探针总数的比例,所以0 ≤Mij≤1。本研究用W∈Rm×k表示每个位点的细胞类型甲基化水平,即特征矩阵,其中Wih∈W;此外,用H∈Rk×n表示k个细胞类型在样本中的比例矩阵,Hhj∈H。根据文献[26],常用的DNA甲基化混合物模型是

其中,误差项εij服从正态分布。式(3)和(4)中的约束要求细胞比例是正的,并在每个样本中的总和为1;式(5)中的约束要求细胞类型的甲基化水平在[0,1]范围内。
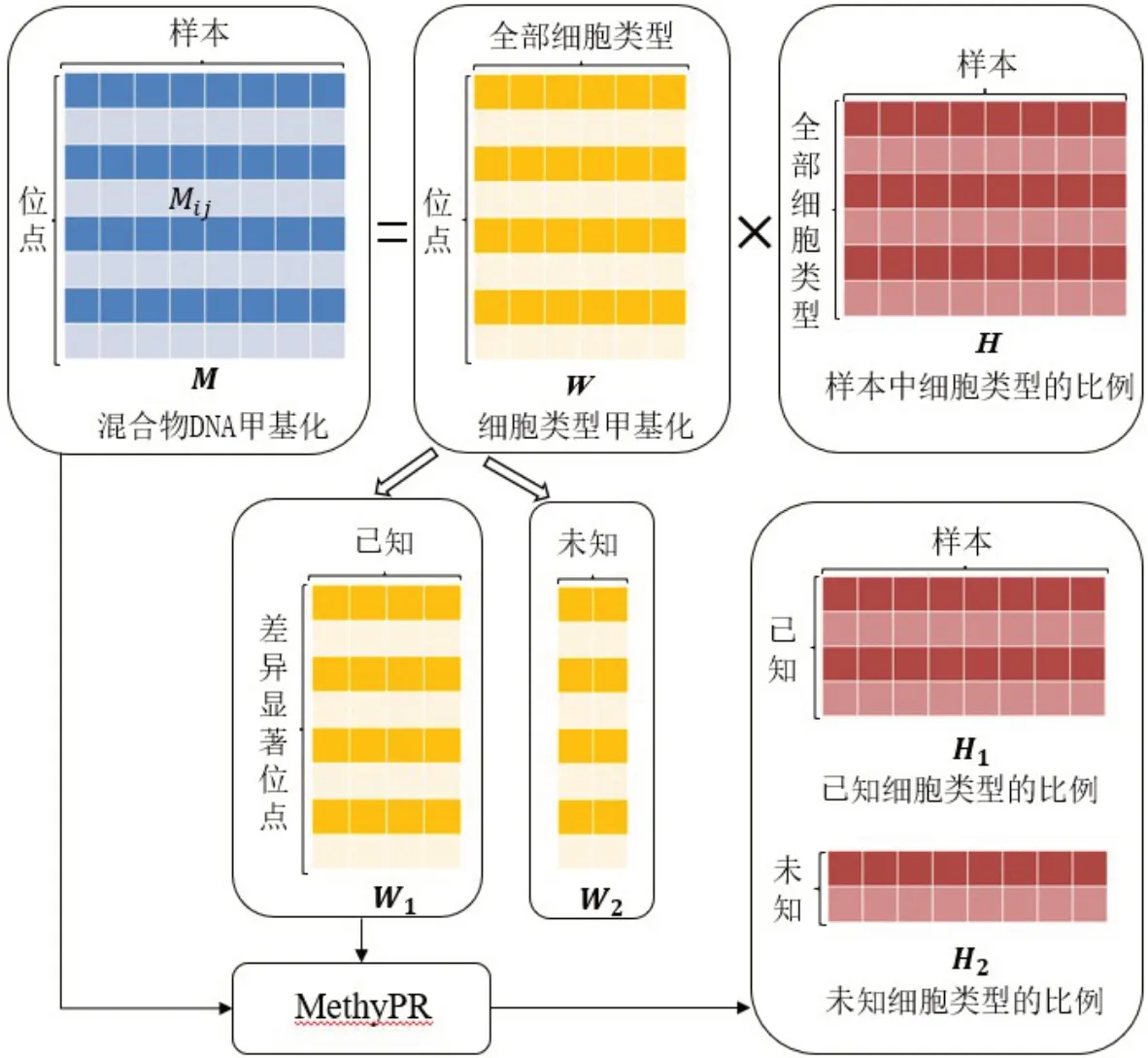
图1 MethyPR模型的框架Figure 1 Framework of MethyPR model
在大多数情况下,矩阵W的数据并不是完全已知,即研究中讨论的细胞类型部分已知的情况。此时模型可以修改为:
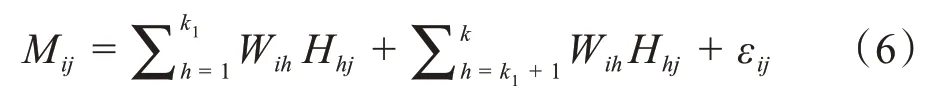
矩阵W分为W1和W2,即式(6)中的Wih(1 ≤h≤k1)和Wih(k1+ 1 ≤h≤k),W=(W1,W2),样本细胞比例矩阵H分为H1和H2,即式(6)中的Hhj(1 ≤h≤k1) 和Hhj(k1+ 1 ≤h≤k),,其中W1和H1是已知的细胞类型所对应的甲基化水平和所占的比例,W2和H2是未知细胞类型所对应的甲基化水平和所占的比例。
给定肿瘤样本的甲基化矩阵M和已知细胞甲基化矩阵W1,目标是得到所有细胞类型对应的比例矩阵H。这个问题可以转化为寻找合适的H1、H2和W2,使全局误差平方和最小:

1.3.2 模型求解采用迭代的非负矩阵分解方法来求解模型,具体流程如下:
(1)随机给定W2矩阵的初始值;
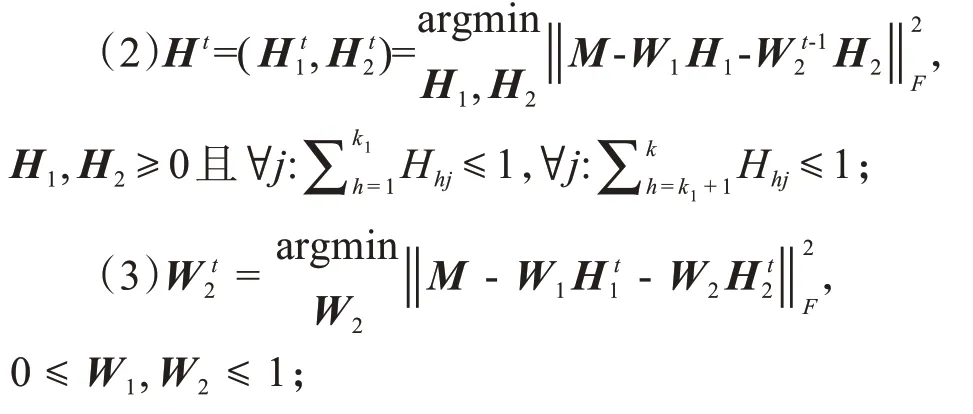
(4)不断重复(2)和(3)的步骤,直至收敛或者达到预定的最大迭代次数。
其中,t为迭代次数。在第(1)步中,使用RPMM 算法[27]来初始化未知细胞类型的甲基化水平。在运用MethyPR时要注意,该方法适用于下载的定量基因组的原始数据,如原始reads 数或CpG 位点甲基化水平,不适合使用logit 变换的数据,因为变换会破坏数据中存在的线性关系。
1.3.3 细胞类型数量确定和模型质量评估采用赤池信息准则(Akaike Information Criterion,AIC)度量来确定肿瘤样本中细胞类型的数量。AIC 是衡量统计模型拟合优良性的一种方法,它不仅考虑了模型的拟合优度,也考虑了当模型成分数量增加时可能发生的过度拟合。由于本实验中的样本量小,因此采用适用于样本量小的改进版本AICc。AICc 的公式为:
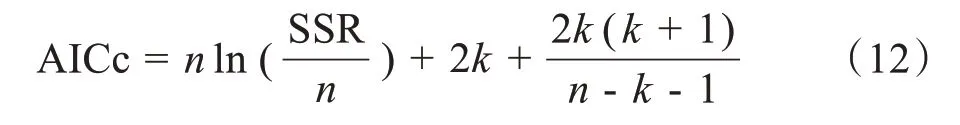
其中,n表示样本量;k表示模型参数的数量(细胞类型的数量);SSR 为残差平方和。不同的k值在模型中对应不同的AICc 值,在所有结果中,AICc 最小时所对应的k就是最优的细胞类型数。
采用两个评价指标来评估MethyPR 的性能,分别为均方根误差(Root Mean Squared Error, RMSE)和Pearson 相关系数。RMSE 是指对预测值与真实值差平方的平均值求平方根,这是回归问题常用的性能指标;Pearson 相关系数用于衡量真实值和预测值之间的相关程度,其值在[-1,1]之间,绝对值越接近1,相关性越强。本研究用模型预测的细胞类型比例和真实细胞比例的Pearson相关系数来衡量评估方法的精度,另一方面,使用真实细胞类型的比例和预测的细胞比例之间的RMSE作为评价指标。
2 结果与分析
2.1 模拟混合物验证
首先,在模拟混合物上对本研究提出的方法进行基准测试,模拟混合物包含6 种纯化的免疫细胞(CD4+T 细胞、CD8+T 细胞、巨噬细胞、B 细胞、单核细胞和粒细胞)和一种乳腺癌癌症细胞(MCF-7)。下载这7种细胞类型的甲基化水平之后,将肿瘤细胞以90%的含量添加到混合物中,剩余部分由6 种免疫细胞类型按随机比例构成。
对于上述基准数据集,将现有的3 种方法(QP[13]、CIBERSORT[6]、EDEC[21])与本研究提出的方法进行对比。QP 是基于参考的方法,直接使用组成细胞类型的参考甲基化谱,将其与混合物样本甲基化谱作为输入,利用二次规划来推断混合物样本中的细胞类型比例。CIBERSORT 内核为支持向量机的一个实例(ν - SVR),将其与细胞类型差异显著的特征矩阵相结合,从而预测细胞类型的比例,其中参数ν给出了训练误差的上界和支持向量的界。EDEC假设混合物样本的对应位点甲基化谱为细胞类型特异性甲基化谱和细胞类型比例的线性组合,随机初始化细胞类型甲基化谱,通过二次规划求解约束最小二乘问题,得到估计的细胞类型比例。
已知有6 种免疫细胞,本研究假设模拟混合物的细胞类型数量为7~15,将细胞数量、模拟混合物数量以及预测结果与模拟结果的误差作为AICc指标的输入,进而推断混合物中细胞类型的总数量。由图2发现,当细胞类型数量为7 时AICc 值最小,最小AICc值识别的细胞类型数量与我们构建模拟混合物时的数量一致。接下来,对于肿瘤含量为90%的混合物,将混合物样本和6 种纯化免疫细胞经过预处理后对应的位点处的甲基化水平作为方法的输入,而乳腺癌细胞的甲基化水平未知,用4种方法分别估计了模拟混合物中6种免疫细胞类型的比例。
如图3 所示,分别计算了4 种方法预测的免疫细胞实际比例与预测比例的RMSE 和Pearson 相关系数。从图3a 可以看出,本研究方法在组成混合物样本的所有免疫细胞中取得最小的RMSE 分别为0.005 4、0.003 3、0.006 4、0.006 0、0.008 4、0.005 6);图3b显示了4 种方法在所有免疫细胞类型上的RMSE 平均值,可以观察到本研究方法明显优于其他方法。图3c 和图3d 为4 种方法在Pearson 相关系数指标下的比较,MethyPR 的性能显著高于其他方法,平均Pearson相关系数指标达到0.97。
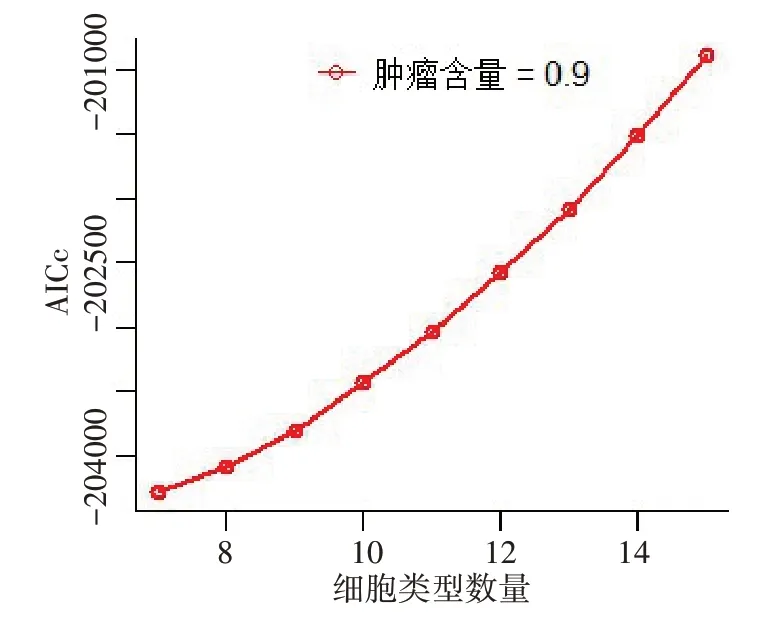
图2 识别模拟混合物中细胞类型数量的AICc线图Figure 2 AICc line graph for identifying the number of cell types in the simulated mixture
为了测试本研究方法对不同噪声水平和生成肿瘤混合物时不同细胞类型含量下的稳健性,进一步进行了以下实验。固定混合物中肿瘤含量为90%,在模拟混合物中加入不同的噪声水平,从0.1 逐渐增加到0.5,比较MethyPR 和其他3 种方法在不同噪声数据上的预测性能,其中每次模拟重复20次。另外,固定噪声水平为0.1,将肿瘤细胞的含量从90%降低到50%(每次减少10%)添加到混合物中,混合物样本中的剩余部分由6种免疫细胞类型按随机比例构成,每次模拟同样重复20 次。在这两个参数的不同设置下,估计6种免疫细胞类型的比例。
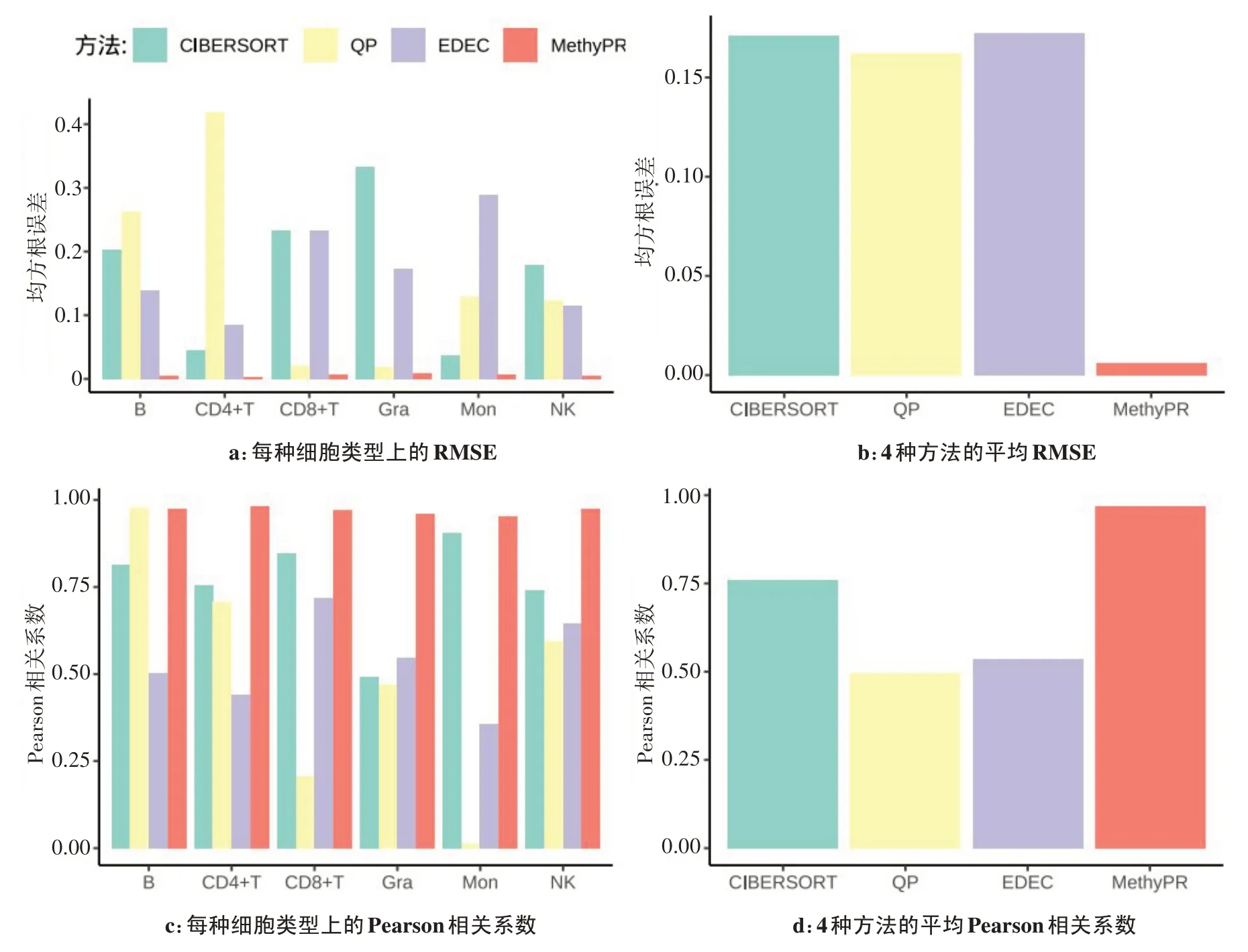
图3 MethyPR与其他算法在细胞水平上的比较Figure 3 Comparison of MethyPR and other algorithms at the cellular level
在肿瘤含量分别为50%、60%、70%、80%、90%的情况下,计算AICc 值。由表1 可知,混合物的细胞类型数量均在7时取得最小,这与构建混合物的数量相一致,说明采用AICc 值预测肿瘤细胞类型数量得到了很好的结果。由表2 可知,在不同的噪声水平下,也得到了类似的结果。
比较4 种方法在不同噪声水平和不同肿瘤细胞含量下的预测性能(图4)。在不同的噪声水平下,得到细胞比例的RMSE 和Pearson 相关系数如图4a 和图4b 所示。MethyPR 在不同噪声水平下均优于其他方法,对于低噪声水平的性能是最佳的,但是在高噪声下,QP 的性能逐渐赶上MethyPR。从图4c 可以看到,MethyPR在不同肿瘤细胞含量下取得稳定的较小RMSE,而其他方法随着未知肿瘤含量的增加,RMSE越来越大;图4d中,随着肿瘤细胞含量的增加,4种方法的预测性能逐渐下降,但在所有情况下,MethyPR具有最高的Pearson相关系数。
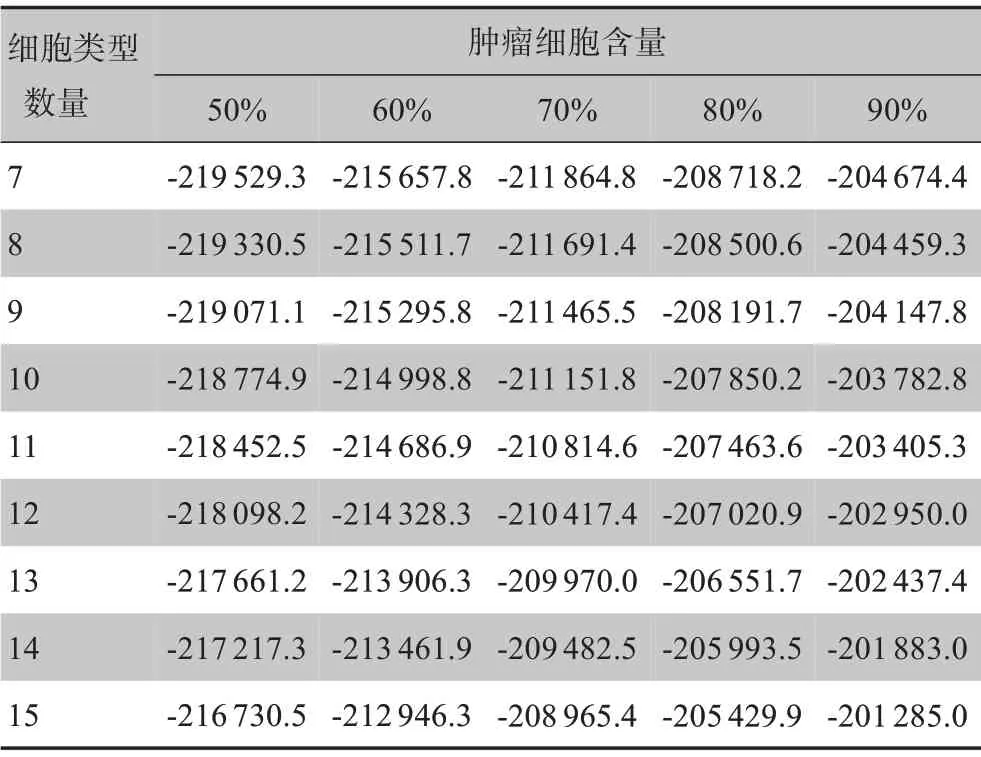
表1 生成混合物中不同肿瘤细胞含量下的AICc值Table1 AICc values of the generated mixture containing different tumor cell contents
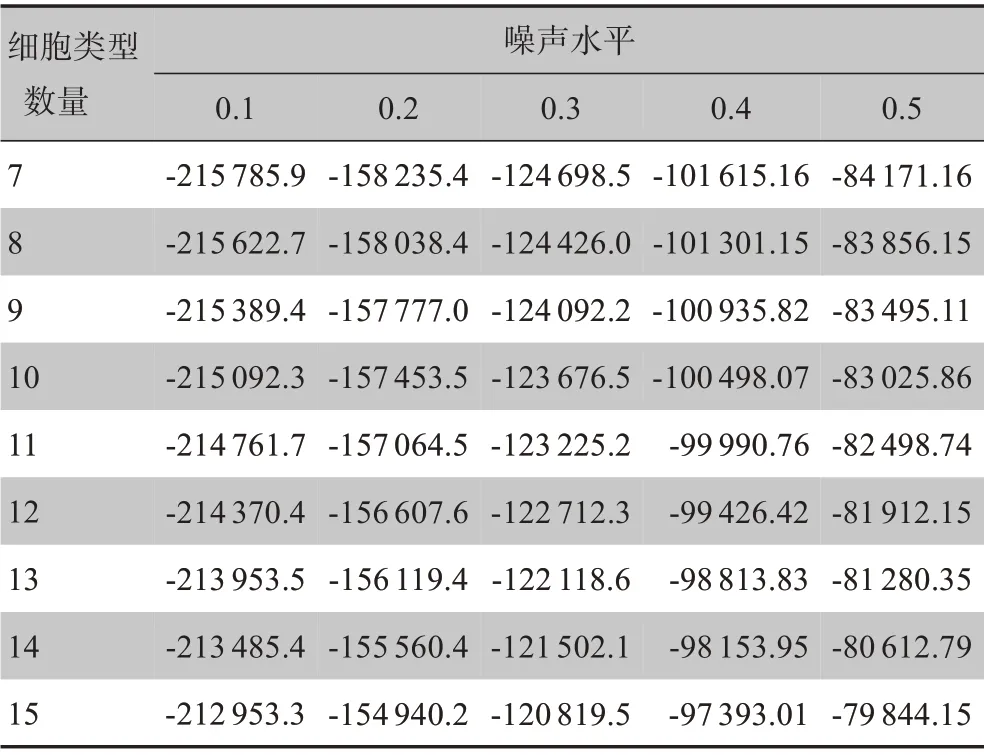
表2 生成混合物在不同噪音水平下的AICc值Table 2 AICc values of the generated mixture at different noise levels
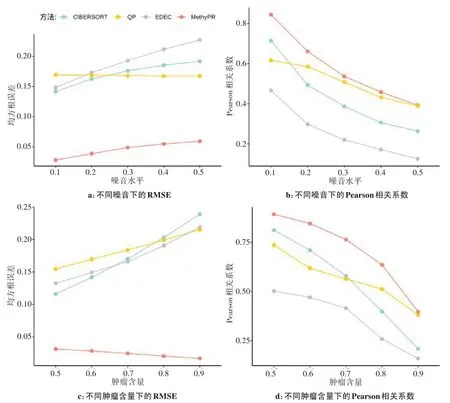
图4 不同方法在不同噪声水平和不同肿瘤细胞含量下的性能评估Figure 4 Performance evaluations of different methods at different noise levels and different tumor cell contents
2.2 体外制备的细胞系混合物验证
在Onuchic 等[21]体外制备的细胞混合物上验证本研究提出的方法。从GEO 下载MCF-7、HMEC、CAF和CD8+T细胞的甲基化谱,由3对组合(MCF-7/HMEC、MCF-7/CD8+T 细胞和MCF-7/CAF)各按两种比例(75%:25%和95%:5%)生成6 个样本,每个样本通过靶向亚硫酸氢盐测序进行分析。采用数据预处理方法得到149 个在不同乳腺癌细胞类型具有显著差异的位点并用于本研究方法。在癌症细胞MCF-7 未知的情况下(图5a),MethyPR 估计的免疫细胞比例和真实值之间有很强的一致性(R=0.996),同时组成混合物的MCF-7 细胞比例与真实值也有很强的一致性(R=0.993)。
临床病理学家根据H&E 染色对29 个乳腺肿瘤样本进行细胞类型组成评估,估计癌性、正常、基质和免疫细胞的比例。本研究假定癌性细胞未知,将其余3 种细胞类型差异显著的特征矩阵和29 个样本的混合甲基化矩阵作为输入,用MethyPR 估计29 个样本中细胞的比例,观察到估计的CD8+T 免疫细胞与真实免疫细胞的比例具有较高的一致性(R=0.81),而癌症细胞的相关性与之相比较低(R=0.71),见图5b。从图5 可以看出,MethyPR 可以准确地预测免疫细胞和剩余其他细胞类型的比例。
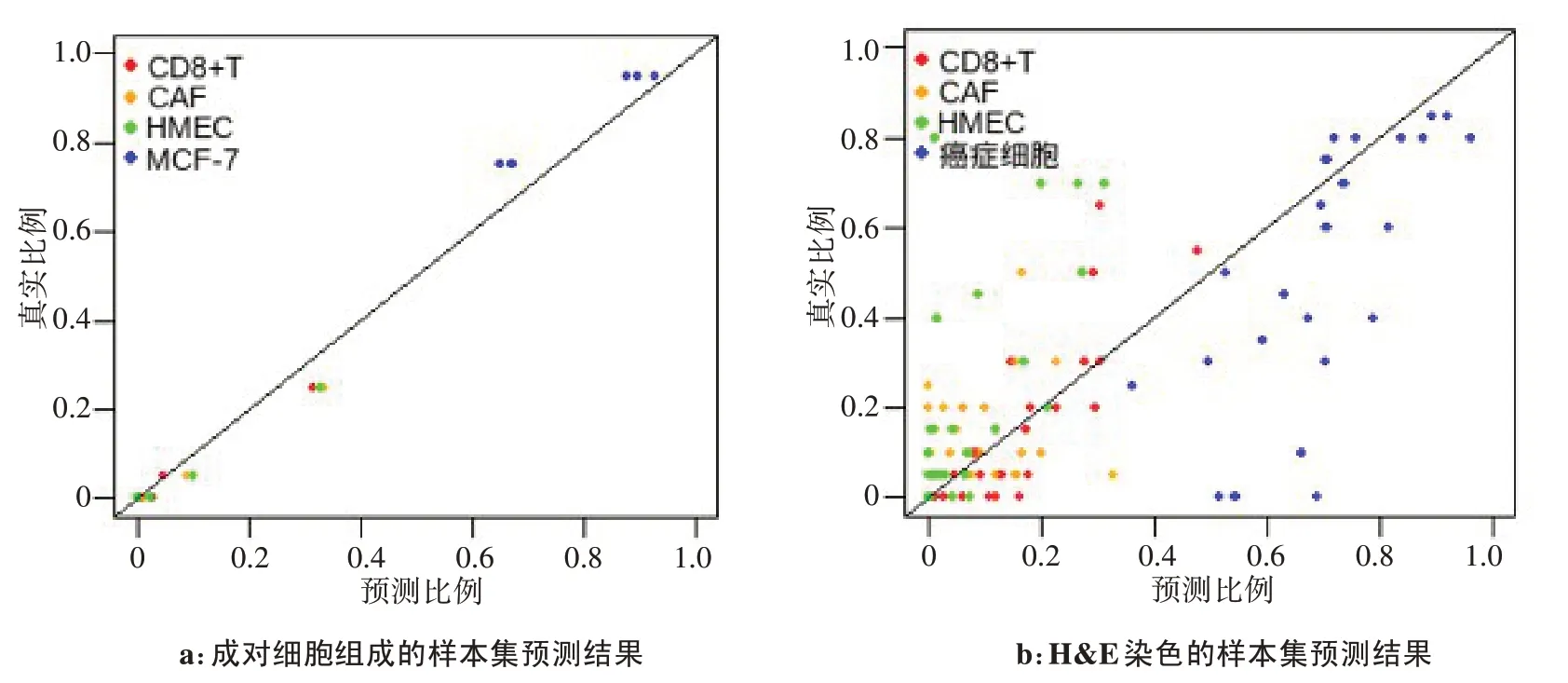
图5 MethyPR在体外制备的混合物上的验证Figure 5 Validation of MethyPR on the mixture prepared in vitro
2.3 肿瘤免疫浸润细胞成分估计
从TCGA的4种肿瘤样本中分别选取了100个样本,然后将MethyPR 应用到这些数据上,估计样本中浸润免疫细胞的比例。如表3所示,不同类型的肿瘤样本表现出不同的免疫细胞浸润模式。幼稚T 细胞在胸腺瘤样本中占有最高的比例(0.031),这与之前的实验研究一致[28]。在结直肠癌样本中,以T细胞为主[29],而急性髓系肿瘤样本具有高比例的单核细胞,弥漫性大B细胞淋巴瘤样本中的B细胞比例最高,这分别与文献[21]和文献[30]结果相一致。
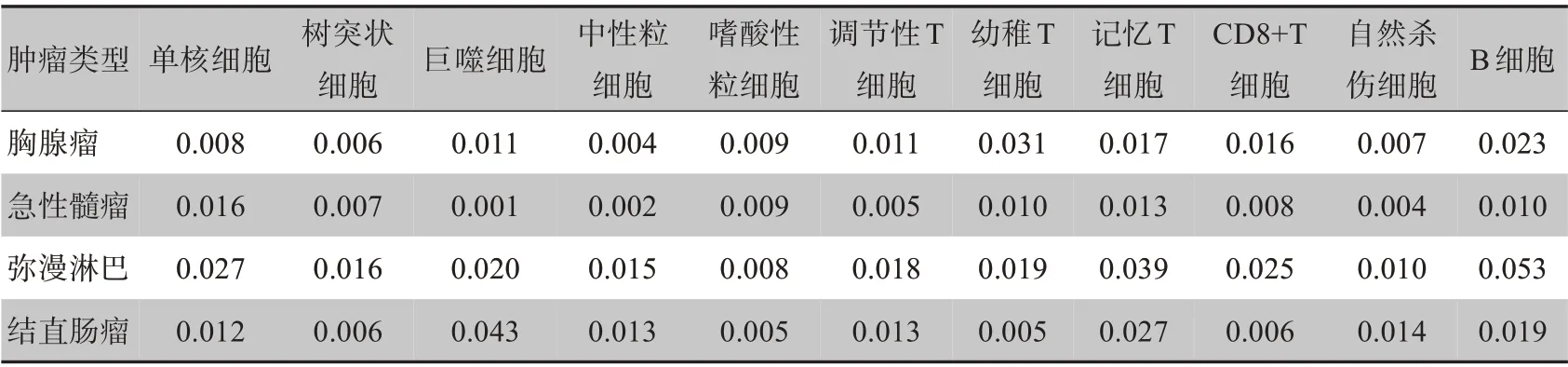
表3 肿瘤浸润免疫细胞在TCGA样本中的比例Table 3 Proportions of tumor-infiltrating immune cells in TCGA samples
3 结论
本研究提出了一种使用甲基化数据对肿瘤微环境进行稳健反卷积的方法MethyPR,该方法基于非负矩阵分解方法,利用容易获得的免疫细胞类型的表观基因组信息,从DNA 甲基化估计细胞组成比例。作为解决基于参考与无参考方法局限性的新方法,MethyPR 能够基于部分参考数据推断混合物中的细胞类型比例。在模拟混合物、实验混合物和真实混合物上,本研究方法都表现出良好的性能,其预测精度高,可以帮助减少估计肿瘤组成的时间和金钱成本,快速获得肿瘤混合物的免疫细胞比例,为表观基因组研究提供了新的思路。
