基于埃博拉病毒糖蛋白单体结构从中药天然产物库中筛选新型抗病毒化合物
2022-05-25史海龙王月雯冯雪松
史海龙,黄 月,程 怡,王月雯,冯雪松,晁 旭
陕西中医药大学,西咸新区 712046
埃博拉病毒(ebola virus,EBOV)是一种包膜丝状病毒,其引起的急性出血热是多发于灵长类动物的人畜共患传染病,其病死率高达50%~90%。近几年非洲西部多次暴发严重的埃博拉疫情,但是迄今为止尚无特效的抗埃博拉出血热治疗药物获准上市。EBOV进入宿主细胞是感染的关键步骤,由病毒表面跨膜单链糖蛋白(single glycoprotein of ebolavirus,EBOV-sGP)介导,这种I 类融合蛋白包含二硫键连接的两个亚基,即表面暴露的GP1与嵌入病毒膜GP2。在通过细胞表面附着因子与宿主细胞结合后,病毒被内吞,内体相应的半胱氨酸蛋白酶、组织蛋白酶切割掉GP1亚基的大部分肽链,裂解暴露受体结合位点[1],这种GP构象的转变,有利于宿主-病毒膜融合和随后病毒基因组的释放。这种融合机制类似HIV-1病毒和流感病毒[2]。因此,开发有效干扰EBOV-sGP构象转变的抑制剂分子,有助于阻断病毒入侵和复制。
2019年英国牛津大学结构生物学系Elizabeth E.Fry及其团队通过使用蛋白表达纯化结晶技术已成功制备EBOV-sGP蛋白,并借助X射线衍射实验测定其三维结构[3](RCSB Protein Data Bank ID:5JQ7),提供了计算机虚拟筛选模型。除此之外,由于天然产物,尤其是传统中药成分库,具有结构独特性与多样性,已被用于治疗各种慢性病和传染病[4,5],同样可用于挖掘抗埃博拉病毒抑制剂。基于以上思考,本研究采用计算机模拟技术,从中药天然产物库中筛选EBOV-sGP抑制剂分子。
1 资料与计算方法
分子对接计算均采用Schrodinger 2018的Glide模块、ADME参数计算采用Schrodinger 2018的QikProp模块,MM/GBSA结合自由能预测[6]采用Schrodinger 2018的Prime MM/GBSA模块,分子动力学模拟及MM/PBSA结合自由能预测[7]均采用Gromacs 2019程序包。采用Pymol与Discovery Studio Visualizer软件绘制受体配体结合模式图。
1.1 靶蛋白EBOV-sGP的预处理与活性口袋的确定
从PDB数据库下载EBOV-sGP结构(PDB ID为5JQ7,配体为toremifene)。使用Schrodinger2018软件的Protein Preparation wizard模块对5JQ7结构进行预处理。以toremifene的空间坐标为参考,使用Receptor Grid Generation模块生成Grid文件,用于基于受体结构的虚拟筛选。参考Elizabeth E.Fry课题组发表的文章[8],选择Asn61、Val66、Asp522、Leu554可作为关键氨基酸残基,并且关注Ala101、Leu515、Tyr517、Gly67、Gly102、Arg64、Thr519、Met548、Ile38、Leu186、Leu63与候选药效分子是否产生疏水相互作用、离子键等,并使用Pymol进行可视化处理,如图1所示。
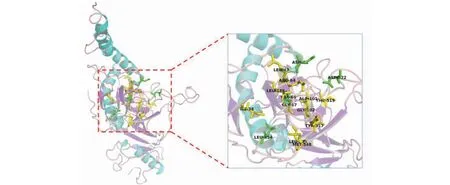
图1 EBOV-sGP活性口袋的空间结构
1.2 中药分子化学结构的预处理
TCMSP数据库[9]中13 144个化合物作为配体库,采用Schrodinger的LigPrep模块对各分子进行加氢、能量最小化、几何优化等处理。在力场OPLS3下,计算pH为7.0±2.0范围的化合物离子状态,搜索合理构象作为对接起始构象,形成可能的互变异构体。以同样方式预处理toremifene。
1.3 基于类药性评估、分子对接模拟、ADME预测、结合自由能计算的多轮虚拟筛选
首先,基于类药性规则“Lipinski Ro5[10]”,“Verber Ro3[11]”对中药分子配体库进行首轮筛选。然后,基于EBOV-sGP活性口袋结构Glide程序对命中分子进行第二轮筛选,依次从高通量模式HTVS-docking、标准模式SP-docking、高精度模式XP-docking进行不同精度的分子对接。接着,基于ADME药代动力学参数进行第三轮筛选,从分子量(molecular weight,MW)、氢键供体(hydrogen bond donor,HB donor)、氢键配体(hydrogen bond acceptor,HB acceptor)、溶剂可及表面积(solvent accessible surface area,SASA)、K+通道阻断相关IC50(IC50value for blockage of HERG K+channels,logHERG)、水溶性(aqueous solubility,logS)、脂水分配系数(octanol/water partition coefficient,logO/W)、口服生物利用度(oral bioavailability,OB)等评估,筛选具有良好类药性质的化合物。最后,基于Prime MM/GBSA结合自由能预测进行第四轮筛选,由于高精度分子对接函数计算仍然无法估算配体-受体复合物结合的溶剂化效应因素,故使用隐性溶剂模型下MM/GBSA算法优化排序。
1.4 分子动力学模拟
为了从动力学和热力学角度动态分析EBOV-sGP与命中分子互作模式,使用GROMACS进行分子动力学模拟。蛋白拓扑参数取自Amber的FF03力场,由PDB2GMX工具获得;抑制剂的各原子电荷先用Gaussian 09在B3LYP/6-311G**基组水平计算静电势,再用Amber中的RESP电荷拟合程序算出。将复合物放入10×10×10 Å3的立方体TIP3P水盒中,并加入Na+和Cl-离子,中和该系统到0.15 mol/L NaCl溶液浓度,随后进行能量最小化和预平衡,最后在300 K温度进行50 ns的分子动力学模拟,时间间隔为2 fs,每10 ps保存一次能量和坐标文件。从35~50 ns稳定模拟轨迹中提取150帧轨迹文件,计算MM/PBSA结合自由能。
2 结果与讨论
2.1 虚拟筛选结果分析
基于靶标EBOV-sGP三维空间结构,从TCMSP数据库13 144个化合物进行多轮虚拟筛选,如图2所示,其具备有较强的靶标结合能、良好的成药性等特质。最终确定打分排名前4的化合物MOL006834、MOL000174、MOL002192、MOL012524,具备有较强靶标结合自由能、良好药代动力学参数、较高生物利用度等特质,结果见表1和表2。随后进行文献检索,发现MOL000174属于中药材苍术的苍术醇类化合物,是动脉炎病毒的强抑制剂[12];MOL006834属于中药材鸭跖草的阿魏酸酰胺类化合物,鸭跖草具有抑菌、抗氧化、抗病毒、降血脂、镇痛消炎的作用,其中阿魏酸酰胺类化合物具有抗炎活性[13];MOL002192属于中药材川芎的阿魏酸酯,具有较好的血管扩张作用及抗菌抗癌活性[14];MOL012524属于中药材牛膝的阿魏酰甲氧基酪胺类化合物,在体外药理实验中显示出有效的抗炎活性[15]。从现有的文献中,4种命中分子具有不同药理活性,但是还没有直接实验证据揭示与本研究靶点的相关性。
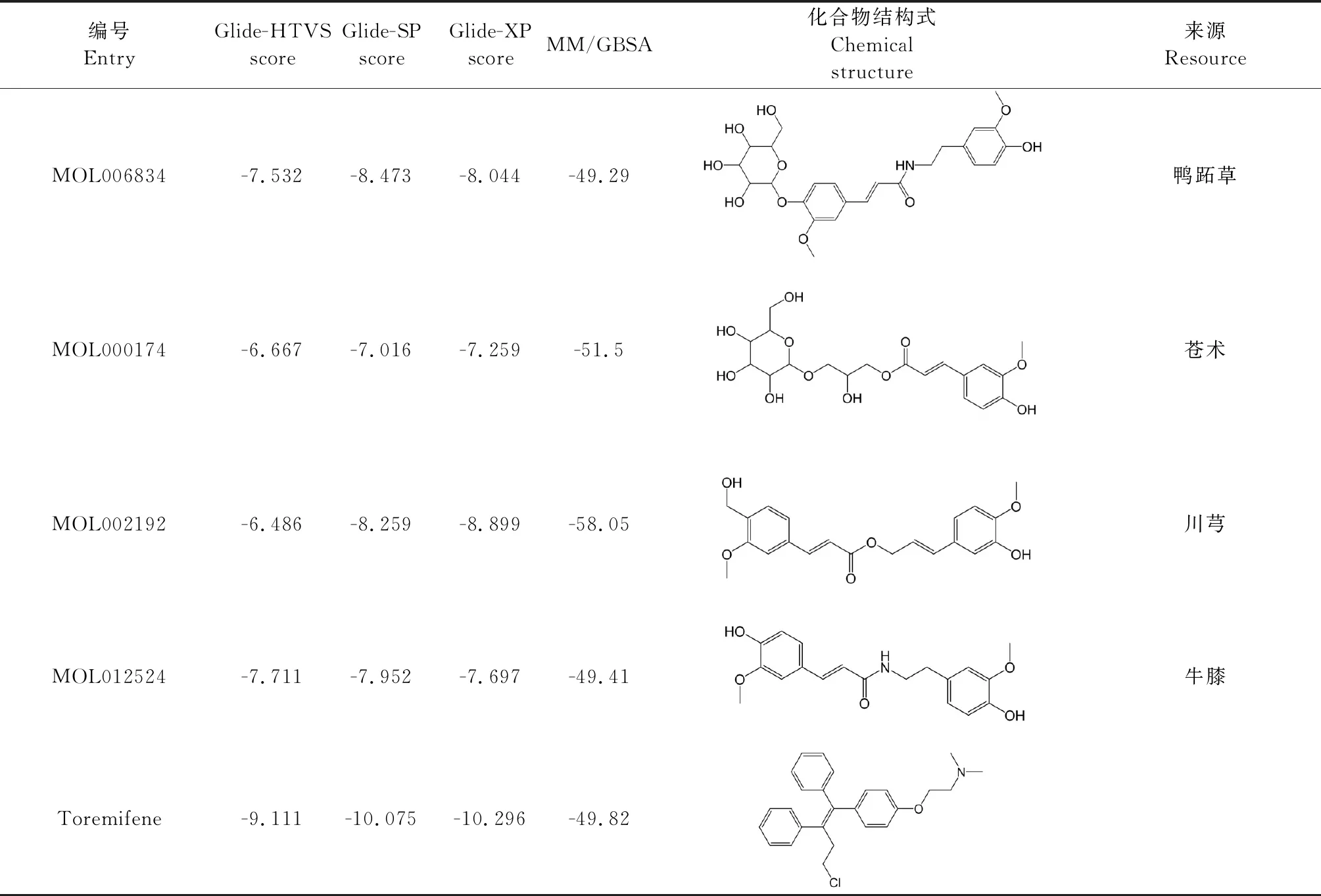
表1 候选化合物及阳性对照Glide分子对接打分值与MM/GBSA结合自由能
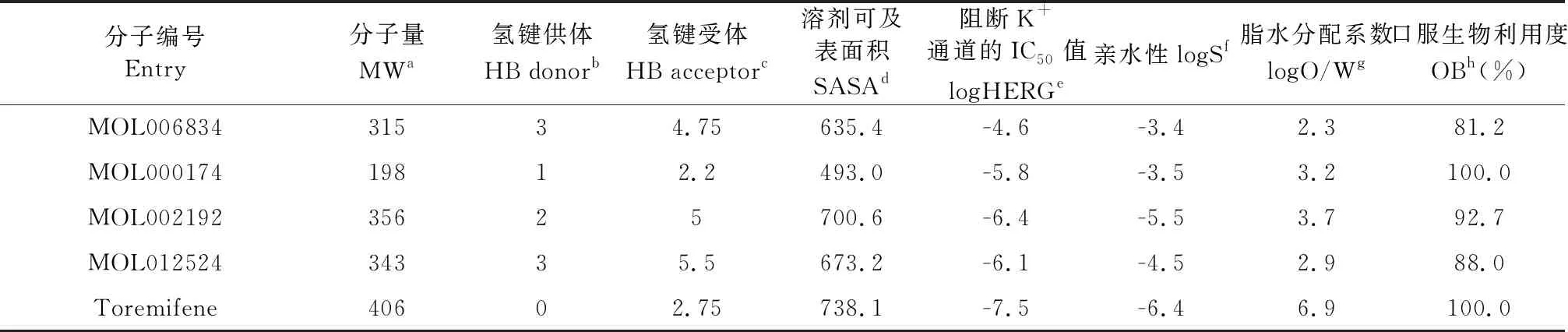
表2 排名前4命中化合物的ADME参数预测值

图2 虚拟筛选流程
2.2 分子动力学模拟结果分析
计算分子动力学模拟50 ns时长轨迹的均方根偏差(root mean square deviation,RMSD),以此检验体系稳定性,结果见图3。在模拟运行35 ns时长之后,所有体系的RMSD的平均值为0.5~0.7 nm,且涨落范围都稳定在0.1 nm以下,均达到了平衡状态。

图3 靶蛋白EBOV-sGP的氨基酸骨架原子随时间变化的RMSD值
通过计算化合物与受体蛋白复合物氨基酸骨架原子在分子动力学模拟过程中的均方根波动值(root mean square fluctuation,RMSF),分析蛋白质中每个氨基酸残基的柔性,评估不同小分子抑制剂对EBOV-sGP每个氨基酸残基波动的影响。发现4个抑制剂复合物体系与toremifene复合物体系都有相似的RMSF数值分布趋势,结果见图4。在铰链区(290~310)、催化loop区(186~201、99~104)、Activationβ-sheet(514~520、62~68)这些结合位点内的残基RMSF都明显较小,说明MOL006834、MOL000174、MOL002192、MOL012524一样都可以与EBOV-sGP结合口袋形成稳定的相互作用,稳定蛋白结合口袋的残基。除此之外,复合物体系(MOL0 06834、MOL000174、MOL002192、MOL012524、toremifene)与靶蛋白空载体(in an inactive or unbound state,APO form)相比较,含配体分子的蛋白二元结构比空蛋白在活性中心关键氨基酸残基520~550区间具有更小的RMSF。

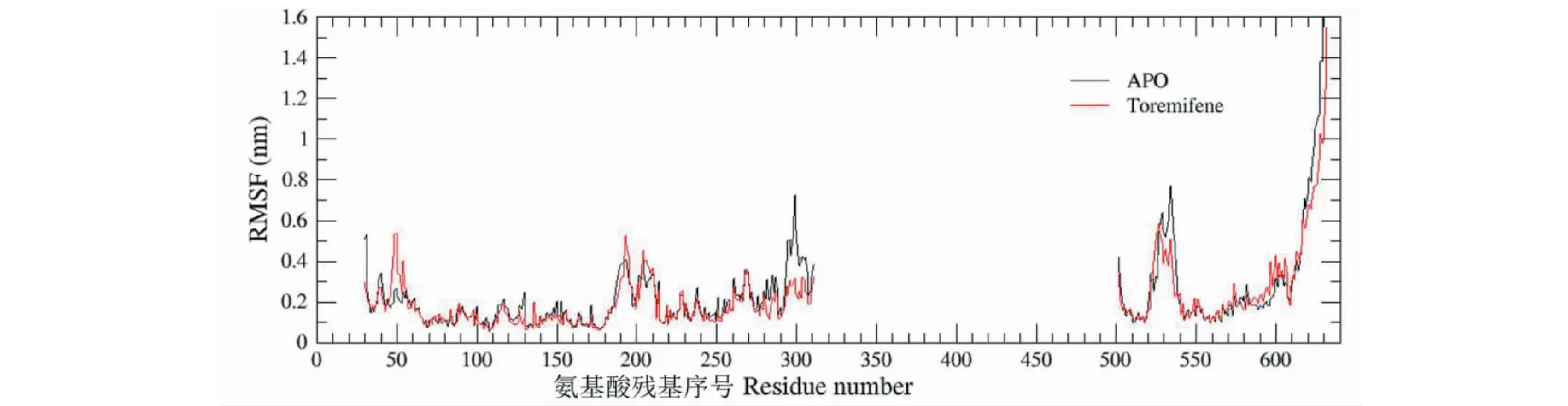
图4 配体-蛋白复合物氨基酸骨架原子的RMSF值波动图
2.3 结合自由能计算结果分析
从35~50 ns分子动力学轨迹中提取150帧轨迹文件,计算MM/PBSA结合自由能及各种能量贡献情况,结果见表3。范德华作用能与静电势能最有利于4种命中化合物与EBOV-sGP结合,表面溶剂化作用也有利于结合,但与范德华作用能和静电势能的数值相比作用较弱,而极性溶剂化作用不利于体系结合。表明范德华作用成分能在整个作用力占有主导地位,范德华作用能成分极大地促进了抑制剂与靶标蛋白的结合,决定了结合自由能的最终排名,这也是MOL006834、MOL000174与靶蛋白EBOV-sGP结合自由能具有优势效应的关键因素。

表3 命中化合物与靶标EBOV-sGP的MM/PBSA结合自由能
MOL006834与MOL000174来源于相同的化学结构母核,空间结构相似,因此范德华作用能、表面溶剂化作用、结合自由能均非常接近;但是MOL000174具有较强的极性溶剂化负效应,具体数值高达205.799 kJ/mol,非常不利于配体与受体结合,因此与蛋白质结合过程中由于溶剂中的极性溶剂化相互作用很大程度上抵消了静电势能与范德华力之和,不利于导致最终的结合自由能排名次于MOL006834,位居第二。
同样,MOL002192与MOL012524也来源于相同的化学结构母核,空间结构相似,因此范德华作用能、表面溶剂化作用、结合自由能均非常接近。MOL012524具有较强的极性溶剂化负效应,具体数值为156.173 kJ/mol,不利于配体与受体结合,虽然静电势能也是-53.094 kJ/mol,但是与蛋白质结合过程中由于溶剂中的极性溶剂化相互作用终究抵消了静电势能与范德华力之和。因此MOL012524具有最弱的范德华作用正效应与较强的极性溶剂化负效应,导致最终的结合自由能不占优势,排名第四。
结合自由能数值越小,表示结合所需能量越低,越有利于配体与蛋白的结合。比较4种化合物与EBOV-sGP的结合自由能,可得出候选抑制剂分子结合靶标强弱关系如下:MOL006834>MOL000174>MOL002192>MOL012524,均优于阳性对照分子toremifene。
2.4 候选化合物与靶标的相互作用位点分析
提取复合物分子动力学模拟50ns稳定后最后一帧构象,观察命中化合物与EBOV-sGP的空间结合构象。由图5可见,靶蛋白EBOV-sGP与MOL006 834之间的相互作用不仅有范德华力、氢键,还有π-Anion相互作用、π-Sulfur相互作用、π-Alkyl相互作用、π-π堆积作用。MOL006834与氨基酸残基Met548、Arg200、Pro533、Pro537、Ala539、Asn204形成常规氢键,并且同样与Glu201、Asn204均形成一个碳氢键。与Ala101、Pro198、Pro202分别形成一个π-Alkyl相互作用,与Glu100形成π-Anion相互作用,与Met548形成π-Sulfur相互作用,与Tyr517形成π-π堆积作用。这些作用力使结合能降低,亲和力增加,候选单体与EBOV-sGP蛋白的活性部位紧密结合,形成稳定的复合物,就可以有效阻止外来病毒粒子附着并侵入宿主细胞。

图5 靶蛋白EBOV-sGP与MOL006834相互作用
由图6可见,靶蛋白EBOV-sGP与MOL000174之间的相互作用不仅有氢键、范德华力,还有π-π堆积作用、π-Alkyl相互作用。MOL000174与氨基酸残基Arg64、Arg164、Arg200、Glu201、Thr520形成常规氢键,并且同样与Gly67、Ala101、Arg200、Thr519、Glu201、Leu199形成碳氢键,还与Tyr517形成π-π堆积作用,与Ala101形成π-Alkyl相互作用。这些作用力使结合能降低,亲和力增加,候选单体与EBOV-sGP活性空腔部位紧密结合,形成稳定的复合物,就可以有效阻止外来病毒粒子附着并侵入宿主细胞。但是还可以观察到氨基酸残基Arg64与Glu100之间存在着不利相互作用,不利于构象稳定。
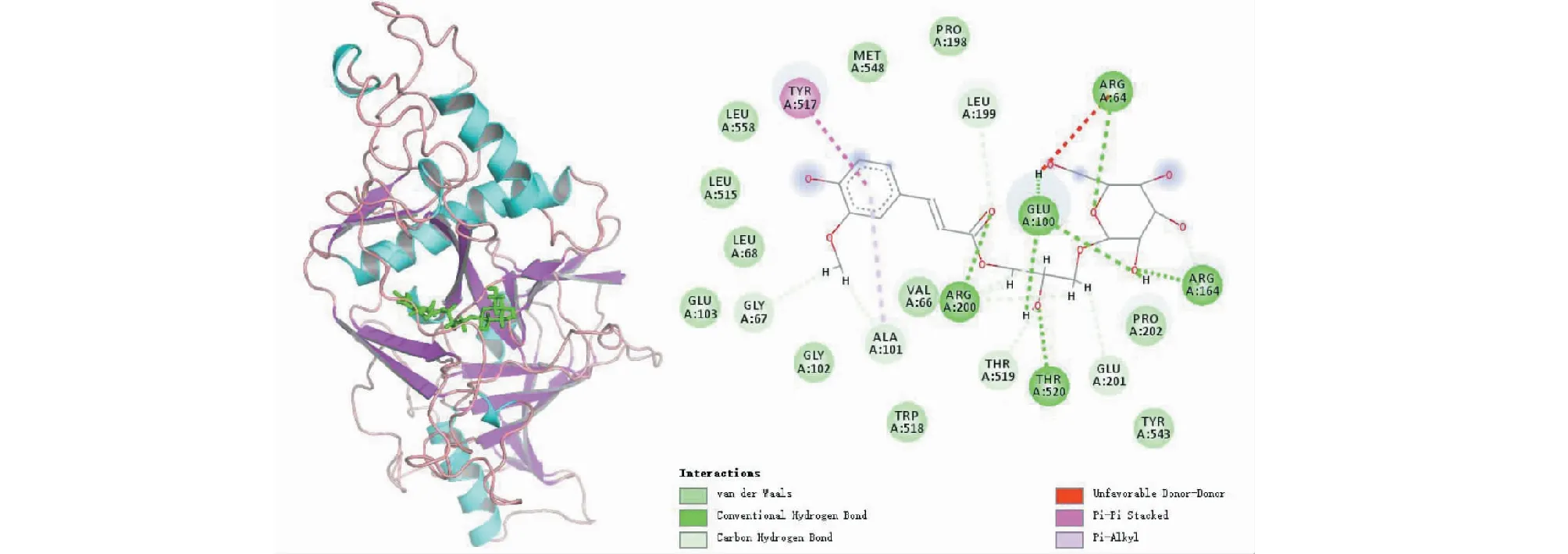
图6 靶蛋白EBOV-sGP与MOL000174相互作用
由图7可知,靶蛋白EBOV-sGP与MOL002192之间的主要作用力是范德华力、氢键、π-cation相互作用、amide-π堆积作用、π-Alkyl相互作用。MOL002192与靶标氨基酸残基Ser195、Asp192、Arg200、Thr520、Asn61、His197均形成氢键,其中Asn61形成了2个常规氢键,Ser195分别形成了常规氢键和碳氢键各1个。与碱性氨基酸残基Arg200、Arg64均形成π-anion相互作用,与氨基酸残基Pro202形成π-Alkyl相互作用,并且His197形成amide- π堆积作用,与这些作用力促进复合物的形成。

图7 靶蛋白EBOV-EBOV-sGP与MOL002192相互作用
由图8可见,靶蛋白EBOV-sGP与MOL012524之间的相互作用不仅有范德华力、氢键,还有π-Alkyl相互作用。MOL012524与氨基酸残基Glu100、Arg200、Glu201、His197形成常规氢键,同样并且与Arg164、Asn98、Tyr99均形成一个碳氢键,与Thr519形成了一个π-Donor氢键,其中Glu100形成了2个常规氢键。与Arg64、Ala101分别形成一个π-Alkyl相互作用。这些作用力使结合能降低,亲和力增加,候选单体与EBOV-sGP蛋白的活性部位紧密结合,形成稳定的复合物,就可以有效阻止外来病毒粒子附着并侵入宿主细胞。
图8 靶蛋白EBOV-sGP与MOL012524相互作用
氢键在生物大分子中起着重要作用,氢键是蛋白质与配体相互作用的主要推动力之一,在稳定蛋白质-配体复合物方面起着不可或缺的作用[16]。Discovery Studio Visualizer生成的结果显示,阳性对照分子toremifene与氨基酸残基Glu100、Thr520形成氢键相互作用,产生的氢键数目为2个;然而4个候选单体均与EBOV-sGP 氨基酸残基Glu201或Asn61形成氢键相互作用,甚至形成双氢键,这些残基均是候选单体的氢键受体,并且常规氢键总数均大于4个(其中氢键数MOL006834为6个,MOL0 00174为7个,MOL002192为6个,MOL012524为5个)。借助g-hbond程序统计氢键数目,计算50 ns模拟过程中EBOV-sGP与抑制剂分子间的氢键数目以及氢键距离或角度的分布。在Gromacs中判断氢键要满足两个条件,距离和角度,g-hbond统计了满足距离限制的所有原子对,并且也同时满足角度限制,即可判定为氢键,统计结果如图9所示。总体而言,与阳性对照分子toremifene相比,命中的候选分子与EBOV-sGP结合氢键数目更加具有优势,均具有更强的相互作用,推测可能具有更高的抑制作用。
图10展示了阳性对照分子toremifene与靶标的结合构象,可以观察到toremifene与EBOV-sGP 氨基酸残基Val66、Glu515均形成π-sigma相互作用,氨基酸残基Ala101同时与配体分子的两个苯环形成π-alkyl相互作用,与碱性氨基酸Arg64形成π-Cation堆积作用,3个Leu氨基酸残基184、186、558同时与配体分子末端氧原子形成alkyl相互作用,这些作用力促进复合物的形成。同样在4个候选单体与靶蛋白的相互作用分析中,MOL006834、MOL002192均与氨基酸残基Pro202形成π-Alkyl相互作用,MOL000174、MOL012524与碱性氨基酸Ala101也同样均形成π-Alkyl相互作用,这些相互作用在候选配体分子与EBOV-sGP单体空腔的残基结合中也起了辅助有利作用。
图10 靶蛋白EBOV-sGP与toremifene相互作用
3 讨论与结论
Ahmad课题组于2017年首次基于EBOV-GP跨膜糖蛋白三聚体空腔的三维结构为靶标,从Mcule化合物库(https://mcule.com/database/)进行大规模虚拟筛选抑制剂,采用Auto Dock Vina与Flex-X两种分子对接软件,并结合网上在线分析工具admetSAR(http://www.admetexp.org)进行ADMET过滤,找到了三个命中分子[17]。从2000年以来,不断有埃博拉病毒疫情暴发,抗病毒药效分子的研发仍是全球医药研究热点。因此本研究为了从中药天然产物库中挖掘抗病毒活性分子,借鉴了此文章的研究思路,靶标从EBOV-GP三聚体改为单体,并重新设计了多轮虚拟筛选的实验方法,筛选策略使用成本更高的算法,尽可能提高预测的精度,降低虚拟筛选假阳性率,以及更好地预测结合构象。除此之外,分子动力学模拟和结合自由能计算也是研究蛋白质与抑制剂相互作用强度的重要工具,采用GROMACS软件进行分子动力学模拟,并使用 MM/PBSA算法预测体系稳定后的结合自由能,这类方法不仅能在原子和分子的层次上给出蛋白质和抑制剂的相互作用细节,而且还能便于阐明蛋白质和抑制剂复合体的结构亲和能关系。
为了有效干扰EBOV-sGP构象转变,本研究基于EBOV-GP单体活性空腔为靶标,从中药天然产物库中挖掘抑制剂分子,有助于阻断宿主-病毒膜融合。首先遵循Lipinski五原则、Verber三原则,过滤掉违反类药性原则的化合物;然后采用Glide软件进行三种不同精度的分子对接计算,即高通量HTVS、标准精度SP、超精密XP等三种模式,逐步缩小候选化合物库的规模;随后采用Schrodinger的QikProp程序预测命中分子ADMET性质,较admetSAR进行过滤策略,新增了肝毒性HERG_IC50、口服生物利用度等两项药代动力学与药物安全性分析的关键指标,并结合Lipinski Ro5、Verber Ro3预测值,做出相对全面的类药性评估;接着在分析靶标蛋白与命中分子结合方式过程中,使用高性能GPU加速技术模拟在真实生理盐水溶剂条件下生物大分子的运动状态,进行了50 ns时长的分子动力学模拟,并使用了MM/GBSA与MM/PBSA两种算法预测结合自由能,提升富集精准度。最后根据XP-Glide 评分、RMSD、RMSF、MM/GBSA 结合能、MM/PBSA结合能、ADME预测,命中分子MOL006834、MOL000174、MOL002192、MOL012524表现出对 EBOV-sGP活性位点显示出极好的结合亲和力和良好的类药性;从原子与分子层次上预测命中分子MOL006834、MOL000174、MOL002192、MOL012524与跨膜糖蛋白的真实结合构象,包括氢键、疏水相互作用、离子键、π-π堆积力等,与关键氨基酸残基显示出极好的相互作用。这四个命中分子均可在商业上购买获得,也可作为EBOV-sGP抑制剂先导化合物进行开发利用,以及药物活性实验验证。需要指出的是,埃博拉病毒具有高致死率和高传播性,相关生物安全要求别极其严格,抗病毒活性测试实验需要在生物安全4级实验室内进行,国内具备此级别的实验室并不多,制约了有关埃博拉病毒的基础研究。因此本研究成果仅局限于药物开发的早期阶段,缺乏抗病毒活性验证、ADMET评估等实验数据支撑,后期需要联合相关的病毒学研究团队,借助专业的病毒学研究平台开展安全有效的生物学验证。
