Cognitive disorder and dementia in type 2 diabetes mellitus
2022-05-19GenaroOrtizMiguelHuertactorGonzlezUsigliErandisTorresnchezDanielaLCDelgadoLaraFermPachecoMoisMarioMirelesRamrezBlancaMGTorresMendozaRoxanaMorenoCihIrmaVelzquezBrizuela
lNTRODUCTlON
Diabetes mellitus type 2 and its complications
According to recent data published by the World Health Organization, diabetes mellitus (DM) affects more than 400 million people worldwide and has a high burden of morbidity and mortality. DM is a multisystemic disease that involves several organs. The complications related to DM are mainly due to a chronic hyperglycemic condition and can be divided into macrovascular and microvascular damage.Macrovascular complications are currently well known and include arteriosclerotic alterations of the large-caliber arteries with progressive reduction in the caliber of the vessels, and the consequential increase in the incidence of myocardial infarctions and cerebrovascular accidents. On the other hand,microvascular complications affect the peripheral circulation and small-caliber vessels, involving the eyes, kidneys, lower extremities, and the central and peripheral nervous system[1]. At the eye level,diabetic retinopathy first causes preproliferative microangiopathy associated with cotton-wool spots and then the formation of microaneurysms and angiogenesis (proliferative microangiopathy), leading to hemorrhages that can produce detachment of the retina itself, causing progressive decrease or acute vision loss and the need to perform various sessions of laser therapy; which, nevertheless, is often not enough, and can lead to progressive, irreversible blindness[2]. At the kidney level, DM leads to alterations in renal function that present with a progressive course according to different stages, which include the appearance, in succession and at different periods of time, of microalbuminuria and proteinuria; and this damage can progress to manifest as chronic renal failure, resulting in uremia, and the need to resort to dialysis[3] (Table 1).

At the level of the nervous system, decompensated DM leads to an alteration in tactile, thermal, and pain sensitivity, and to the transmission of internal muscular impulses from movement. Additionally,peripheral neuropathy can lead to more or less serious lesions of the foot at the beginning, and to paresthesia or a burning or stinging sensation in the extremities[4], until it leads to malfunction of thevascular and nervous components, and the appearance of ulcers which can complicate with infections,degenerate into gangrene, and cause severe tissue damage that may require amputation[5].
They cut Cassim s body into four quarters, and nailed them up inside the cave, in order to frighten anyone who should venture in, and went away in search of more treasure
However, what is emerging lately is that DM is also associated with another type of very serious and debilitating complication: dementia. In fact, DM appears to be a risk factor for the development of progressive cognitive impairment as well as vascular-based dementia and Alzheimer’s disease (AD)[6].Several studies have shown that people with type 2 DM (DM-2) have a risk factor for mild cognitive impairment and dementia that is greater than 1.5 times. The evidence was strengthened with the early stages of AD that are characterized by an alteration of energy metabolism, and, in particular, by a modified use of glucose[7]. What has emerged from the various studies is that it is not so much the DM itself that causes dementia, but the alterations in blood glucose levels associated with it; as, in fact, there is a close correlation between the glycated hemoglobin value and the possibility of developing dementia. In particular, the The Action to Control Cardiovascular Risk in Diabetes-Memory in Diabetes trial study (ACCORD-MIND) evaluated the relationship between the level of glycated hemoglobin and fasting blood glucose on the ability to perform four cognitive tests that included: the digit and symbol substitution test[8], the Mini Mental Status Examination, the King Auditory Verbal Learning Test, and the Stroop Test. The data showed that a 1% increase in glycated hemoglobin is associated with a significant reduction in scores on various cognitive function tests, and concluded that elevated glycated hemoglobin levels relates to lower cognitive function. However, unlike glycated hemoglobin, a high fasting blood sugar level was not correlated with a worsening of the test score, which shows that chronic hyperglycemia creates the damage, not a single glycemic spike. Nonetheless, what seems worrisome is the demonstration that excessively high blood glucose values are not necessary in order to have this complication[9].
Glycated hemoglobin levels equal to 7% are a value that many diabetologists consider discretely high,and, in and of themselves, sufficient to cause harm. Several mechanisms have been proposed to explain how decompensated DM can lead to dementia: a first hypothesis is that since elevated glucose levels are associated with a higher prevalence of cardiovascular risk factors and cardiovascular disease, the relationship with dementia could be mediated by the latter; however, this hypothesis is refuted by the fact that this relationship is not attenuated by a reduction in cardiovascular risk. It has also been proposed that chronic exposure of the brain to elevated blood sugar levels can accelerate cognitive decline[10]. Additionally, several post-mortem studies in the brains of people with AD have shown the presence of metabolic oxidation products associated with hyperglycemia. Another possibility is related to the fact that high levels of glycosylated hemoglobin imply an inefficient action or a reduced effect of insulin (more on this later) due to insufficient secretion or reduced activity, or both. There are many insulin receptors (IRs) in the brain, some of which play a role in glucose transport, but many are believed to have a role in cognitive and neurotrophic processes. On this basis, therefore, it is suggested that cognitive impairment is a consequence of a reduced action of insulin in the brain[11]. Other studies suggest that frequent hypoglycemia, which the diabetic patient often suffers, may be the cause of dementia: in a recent and large longitudinal cohort study carried out in elderly patients, the presence of severe hypoglycemic episodes was associated with a 2.4% higher risk per year of developing dementia than in DM-2 without this complication[12]. The mechanism by which this occurs is quite intuitive:hypoglycemia, indeed, seems to directly cause neuronal death, especially in particular areas of the brain that are more vulnerable, such as the hippocampus. It should also be taken into account that the prevalence of dementia is higher not only in diabetics but also in hypertensive and dyslipidemic patients, and in many cases these three mechanisms are enhanced in patients with metabolic syndromes. Treatment with antihypertensive drugs in subjects older than 60 years with isolated systolic hypertension, has been shown to reduce the incidence of dementia. The study data would indicate a 50% reduction in the frequency of dementia from 7.7 to 3.8 cases per 100 patients treated per year (
=0.05)[13]. Nevertheless, with respect to dyslipidemic subjects, statin intake would not affect the prevalence of dementia. This is what emerged from the results of the PROspective Study of Pravastatin in the Elderly at Risk study (PROSPER), where it was observed that cognitive function declined at the same rate in both study treatment groups, and no significant differences were observed between the patients in the pravastatin group or the placebo group[14]. Mild cognitive impairment is a risk factor for developing a major cognitive disorder over time, and vascular lesions associated with DM,hypertension, and dyslipidemia can gradually trigger neurodegenerative mechanisms of damage[15].Major cognitive impairment is a highly disabling medical condition that affects all aspects of patient life and the family environment; and, if we take into account that often the diabetic patient is also hypertensive and dyslipidemic, we understand how the risk of developing dementia is very high in these subjects, especially if the risk factors are not well controlled[16].
Yes, I had the “hots” for him and would goggle2(,) eye him playing his guitar/singing while I was on the dance floor, or just standing3 at the bar listening and staring
DM-2 AND DEMENTlA: “A COMPLlCATED AND DANGEROUS RELATlONSHlP”
Although DM affects all age groups, its prevalence is the highest in elderly patients (under 65 years of age): 12% to 25%. Population projections suggest that these rates will double over the next twenty years.The prevalence of dementia, of all etiologies combined, increases exponentially with age, and so it represents a major public health problem with a high probability of worsening in the near future.Therefore, there is a bidirectional association between DM and dementia, each of which increases the risk of “looking” at the other, although we do not know precisely how.
DM-2 AND THE RlSK OF DEVELOPlNG DEMENTlA
A growing number of studies indicate that patients with DM-2 are between 1.5 and 3 times more likely to develop AD or vascular dementia, and this risk seems especially increased in the group of elderly diabetic patients who have a history of severe hypoglycemia during hospital stays or outpatient management, particularly during multiple hypoglycemic episodes[17]. But, is this increased risk of developing dementia in the presence of DM the result of a causal link? As a matter of fact, crosssectional and longitudinal studies using brain MRI show an association between DM and the development of brain atrophy, particularly at the level of the hippocampus and the amygdala. These studies also reveal a link between DM and ischemic strokes (cerebrovascular accident) as cortical and subcortical microinfarcts; and, moreover, severe hypoglycemia leads to brain damage, particularly in the cortex and hippocampus[18]. The pathophysiological hypotheses proposed to explain the increased risk of dementia in the presence of DM are diverse. Microinfarctions can result from hypoglycemia or microvascular changes secondary to hyperglycemia, and are one of several factors involved in this pathophysiology[19]. The latter can also lead to changes in key proteins by glycosylation; and,alterations in intracerebral insulin signaling pathways could cause a loss of cellular ionic homeostasis,oxidative stress, an increase in β-amyloid deposits, and phosphorylation of tau proteins (to be discussed later). This last mechanism is cited more frequently to explain the increased risk of dementia, even in the presence of a pathology compatible with AD[20]. Also, DM-2 continues to be an important cardiovascular and cerebrovascular risk factor that increases the risk of stroke and, ultimately, of vascular or mixed dementia, with an overlapping presentation of both being frequent (15% to 20% of dementias)[21](Figure 1).
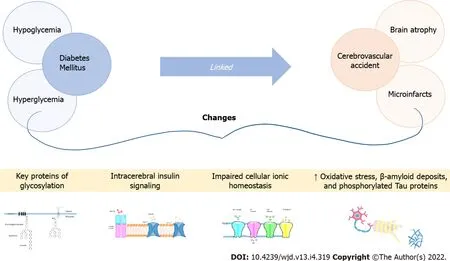
VASCULAR DEMENTlA
First of all, dementia is defined as a decrease in cognitive functions (attention, memory, judgment,
)that leads to a significant loss of autonomy in the affected person; where cognitive impairment and loss of autonomy exceed what is expected in normal aging. The term ‘vascular dementia’ is used when cognitive and functional losses are related to damage to the cerebral vascular network, in other words damage to the vessels that circulate blood in the brain[22]. Keep in mind that we do not always talk about dementia when there is a cognitive disorder. Instead, when the cognitive difficulties are mild, and the autonomy of the person is not compromised, we use the term ‘mild cognitive disorder of vascular origin’ according to the DSM-V. Mild vascular cognitive disorder can progress to vascular dementia(major cognitive impairment) but it does not necessarily do so; thus, the importance of understanding what is happening in order to intervene appropriately[23].
DlFFERENCES BETWEEN VASCULAR DEMENTlA, AD, AND MlXED DEMENTlA
These results suggest that insulin resistance accelerates the onset and increases severity of AD, particularly in situations that predispose to the development of tau disease. Furthermore, the increase in the cytosol of IRS-1 pS312 and pS616 is consistent with the presence of neurofibrillary degeneration in the brains of AD patients, whereas pS312 is limited to the nuclear region of cells in controls. These findings suggest that phosphorylated IRS-1 species may cause tau pathology in AD, beyond their role in the development of brain insulin resistance[69].
She also deprived them of their kingdom, for they had really shown themselves unfit to manage it, and bestowed it upon the Prince and Princess, who, though they were unwilling52 to take it, had no choice but to obey the Fairy

CAUSES OF VASCULAR DEMENTlA
The brain feeds on oxygen and glucose transported in the blood by the cerebral vascular network, which is formed by a multitude of blood vessels that cover all parts of the brain; and the malfunction of these vessels can have the effect of depriving oxygen to specific areas of the brain, therefore weakening or destroying neurons in these areas[27]. Various problems can affect the vascular network and its many blood vessels, such as the narrowing, blockage, or rupture of arteries or veins. In these cases, the blood flow that ensures the survival of the cells is disturbed and the health of the latter is seen threatened. The involvement of the cerebral vascular network is also usually influenced and accompanied by other diseases like atherosclerosis, DM, or arterial hypertension[22,28].
At radio station WJLT in Fort13 Wayne, Indiana, letter poured in for the Christmas wish contest. The workers had fun reading about all the different presents that boys and girls from across the city wanted for Christmas.
COGNlTlVE DlSORDERS ASSOClATED WlTH VASCULAR ALTERATlONS
The cognitive disorders found in people with vascular damage vary depending on the nature of the problem (narrowing, obstruction, bleeding), the type of vessels affected (small or large vessels), and the location in the brain (forward or backward); on the surface or in the center, and on the right side or on the left side. Therefore, the symptoms can be very varied and affect both cognitive functions (attention,reasoning, language,
) as well as motor and sensory functions[22,29]. Nevertheless, some disorders in diseases of a vascular nature are more common than others. Among the most common symptoms foundare slower processing of information, difficulty with cognitive flexibility, or frequent need for help to remember the information learned. These disorders are also associated with changes in mood, slowed motor skills, increased fatigue, and sometimes a less confident gait associated with postural instability[23]. Some people present a dysexecutive problem with reduced speed of the thought process, attention,working memory, the ability to solve problems and make decisions, making mistakes in terms of daily activities with problems in performing simultaneous, dual or multi tasks, and sometimes failure to remember recent information[30].
The bridegroom was astonished, and thought, She is like my Maid Maleen, and I should believe that it was she herself, but she has long been shut up in the tower, or dead
DlABETES AND AD EPlDEMlOLOGY
Longitudinal studies have shown that the cognitive decline in patients with DM-2 is up to two times faster than in physiological aging, and that diabetic patients are at increased risk of mild cognitive impairment[31,32]. In addition, a pioneering study in the 1990s, the Rotterdam study, investigated the link between DM-2 and different types of dementia including AD. They showed that DM-2 nearly doubled the risk of dementia, with the strongest relationship being to vascular dementia but also observed with AD[33]. The relationship between the accumulation of vascular risk factors (DM, high blood pressure, heart disease, and smoking) and AD showed that DM and smoking were the most important risk factors, and that AD risk associated with DM, regardless of other vascular risk factors,was higher than previously reported[34]. Since then, numerous longitudinal studies have been conducted. Most have identified DM as a risk factor for AD[35]. Studies that specifically analyze the incidence of dementia in DM-2, after adjusting for glycemic control, microvascular complications,comorbidities, and high blood pressure and cerebrovascular accidents, have also shown to have a higher risk. Eight of the thirteen population-based longitudinal studies analyzed found an increased risk of AD in adults with DM, ranging from 50% to 100%[19]. These results were confirmed by two large population studies with a 10-year follow-up[36,37]. Poor glycemic control and duration of DM were later identified as risk factors for AD[38-40]. In a meta-analysis, with a total of 6184 people with DM and 38,530 non-diabetics, the relative risk of AD for people with DM was 1.5 (95% confidence interval: 1.2-1.8)[41]. A meta-analysis involving 1746777 subjects found similar results with a relative risk of AD in DM of 1.53 (95% confidence interval: 1.42-1.63)[42]. Other studies have linked various forms of peripheral insulin resistance[43], such as prediabetes[44], metabolic syndrome[45], obesity induced by a high-fat diet[46], and non-alcoholic fatty liver disease[47], with AD and cognitive impairment (Table 3).
PATHOPHYSlOLOGY
Neurodegenerative diseases such as AD are predominantly sporadic, and the underlying pathophysiological mechanisms are not yet fully understood. Although there are obvious differences between AD and DM, they share pathophysiological mechanisms such as: protein aggregation,mitochondrial dysfunction, chronic inflammatory response, apoptosis, synaptic failure, and decreased neurogenesis. As previously mentioned, numerous epidemiological studies have shown a link between neurodegenerative diseases and DM-2, but the pathophysiological link that unites them remains to be discovered. Even if the vascular hypothesis is widely suggested to explain the possible influence of DM-2 in AD[48-50], the hypothesis most studied at present is that of the implication of the insulin signaling defect which is a fundamental characteristic of DM-2.
There is evidence in favor of the implication of a defect in insulin/insulin-like growth factor 1 (IGF-1)signaling in AD[51,52]. As indicated below, altered insulin signaling influences the pathogenesis of DM-2 and AD. This shows its role in neurodegenerative processes and the probable implication of other pathophysiological mechanisms common to metabolic disorders and neurodegeneration.
THE lNSULlN/lGF-1 SlGNALlNG PATHWAY
Insulin and the IGF-1 are polypeptidic hormones that are very similar in structure and function. Insulin is mainly secreted by the pancreas when blood sugar is perceived to be high, while IGF-1 is mainly secreted by the liver[53,54]. However, these two molecules are also found in the brain[55]. Historically,insulin is considered a peripherally-secreted anabolic hormone that plays a role in the storage and use of metabolic reserves. Insulin also exerts pleiotropic functions on protein metabolism (increased synthesis and inhibition of proteolysis), growth, control of apoptosis, and development. The most studied role of insulin is that it plays a part in glucose homeostasis and energy balance. These peripheral actions are controlled (at the brain level) by neurons, known as glucose-sensitive, that are present in the hypothalamus. Hence, insulin regulates body weight, energy homeostasis, and peripheral lipid and glucose and protein metabolism. Therefore, a defect in insulin secretion or signaling at the periphery or at the central level could cause changes in energy metabolism throughout the body, including the brain[54].
The source of insulin in the brain is still under discussion. It is thought that insulin from pancreatic β cells is transported to the brain through the blood-brain barrier (BBB) by a saturable process
receptors. However, insulin and IGF-1 are also produced by pyramidal neurons in the cortex,hippocampus, and olfactory bulb. Unlike peripheral tissues, brain insulin does not have a direct influence on glucose uptake in neurons, but plays a central role in modulating many functions in the brain and, in general, through effectors posterior, promotes cell survival (Figure 2).
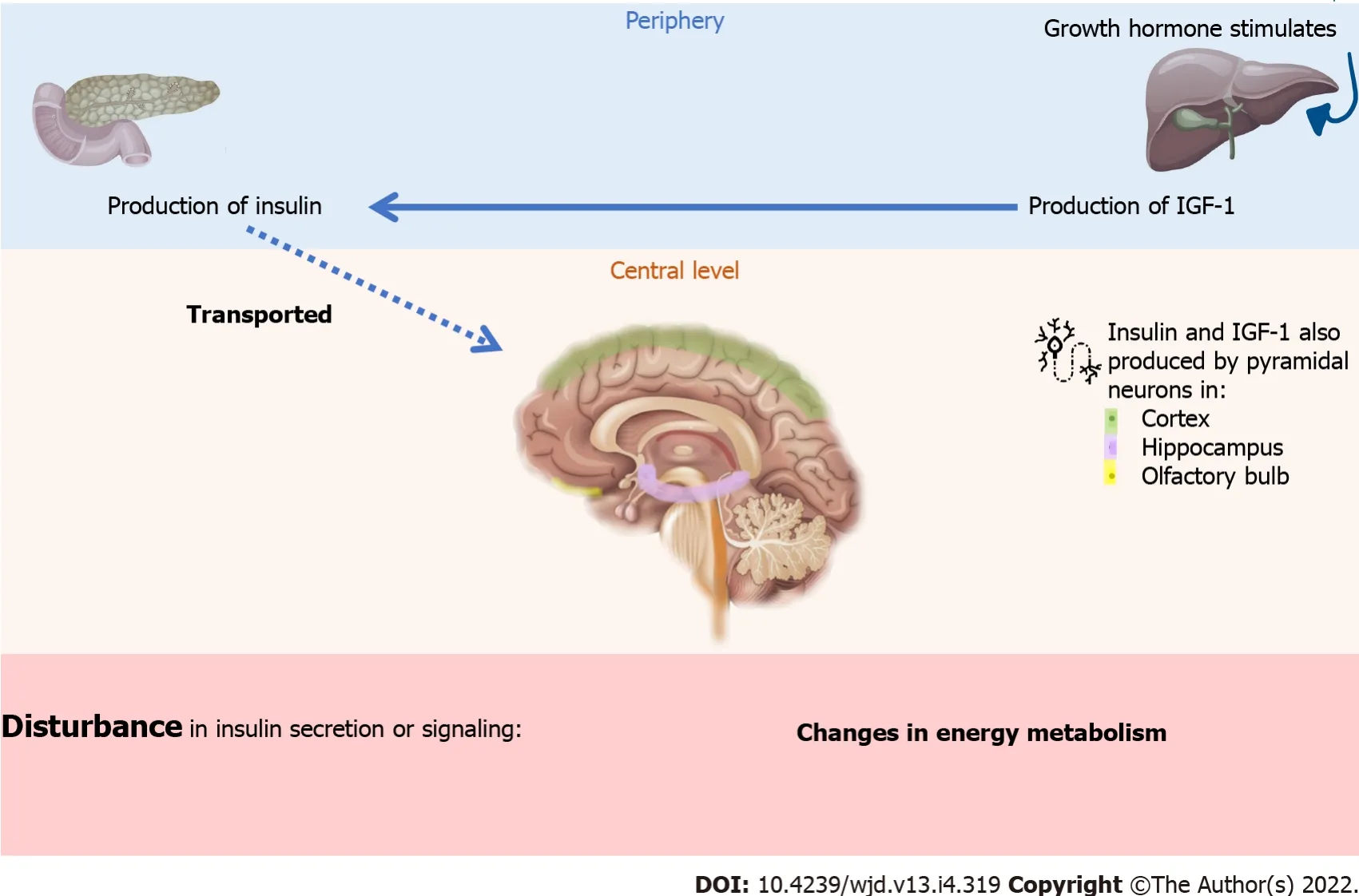
Or, we should consider insulin resistance diseases as processes that can affect one or more organs and tissues, in the same way that atherosclerosis can affect one or more vessels, and produce different manifestations of the disease. On the other hand, there are mechanisms of oxidative stress and chronic inflammation that are associated with clinical processes like DM[49].
I shall take good care to keep by your side, and when the right fish comes I will give you a little push, and with that you will seize the fish and cut it up
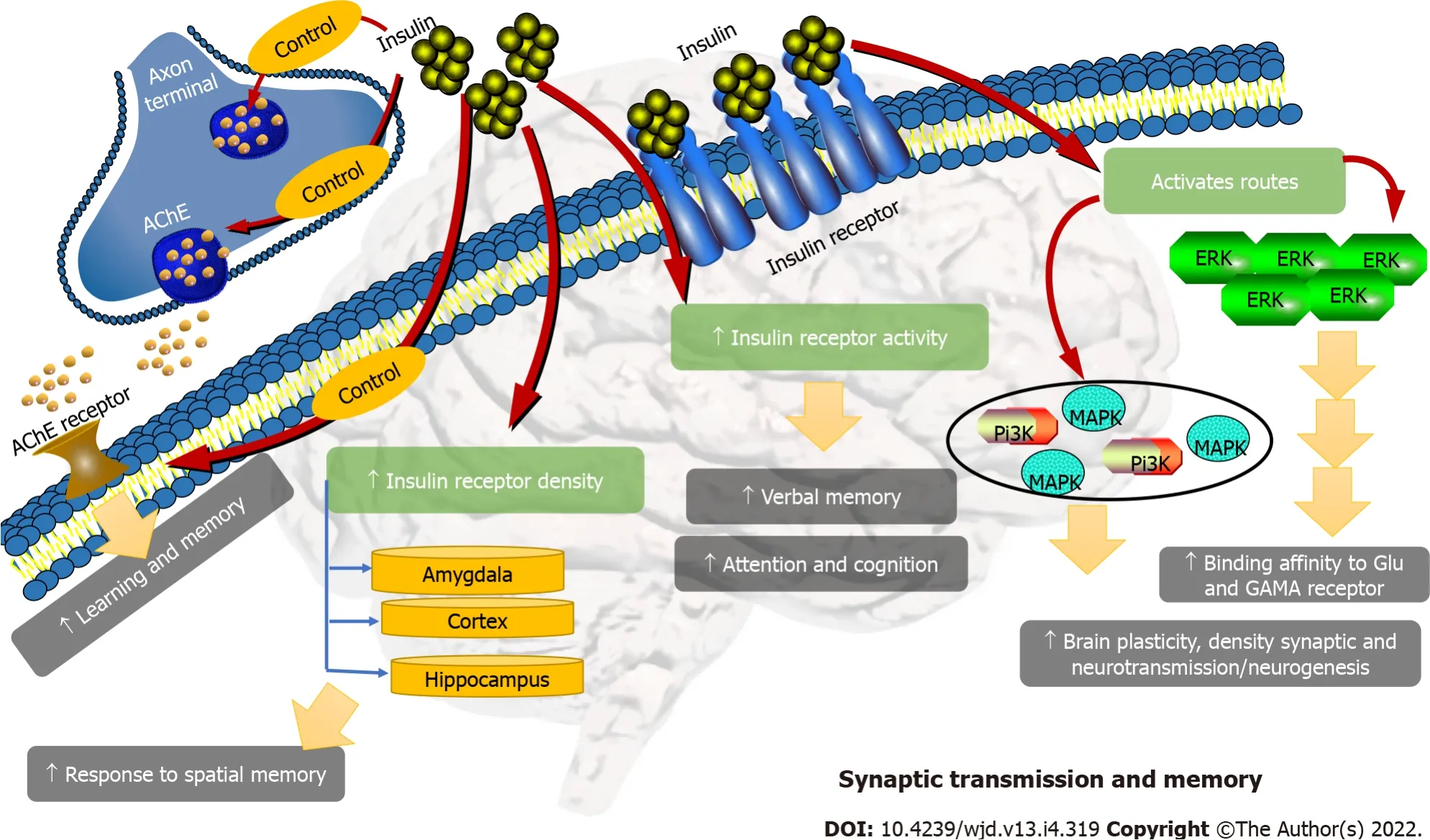
The effects of insulin on cognition appear to depend on the activation of the PI3K/MAPK pathways,through their ability to modulate synaptic plasticity, synaptic density, neurotransmission, and possibly neurogenesis. In addition, insulin induces or modulates a series of neurotransmitters that are part of learning and memory, such as acetylcholine. Activation of the PI3K/MAPK/ERK pathways also serves the effect of glutamate and g-aminobutyric acid receptors. They also enhance protein synthesis and maintain dendritic spine stabilization, which has been shown to be essential for long-term potentiation(LTP) in the hippocampus, and for memory consolidation[74] (Figure 5).
Other mechanisms are also involved in the negative regulation of the insulin signal: dephosphorylation of the phosphoinositides by lipid phosphatases such as PTEN (homolog of phosphatase and tensin deleted on chromosome ten) and SHIP (Src homology 2 containing inositol 5-phosphatase domain) invert the PI3 kinase signal. The PI3K/AKT pathway can modulate several downstream effectors including glycogen synthase kinase-3β (GSK-3β), mTOR (mammalian target of rapamycin)kinase, Caspase-9, and the FoxO1 transcription factor (Forkhead box protein O1). These different posterior effectors regulate a variety of important functions that are commonly disrupted in neurodegenerative disease such as apoptosis, autophagy, inflammation; nerve cell metabolism, protein synthesis, and synaptic plasticity. So, it is not surprising that insulin signaling has finally been shown to improve neuronal survival[59,60].
ROLE OF THE lNSULlN/lGF-1 SlGNALlNG DEFECT
With normal aging, there is a gradual loss of regulation of insulin secretion leading to hyperinsulinism as well as decreased expression and function of IRs. Decreased insulin expression, reduced delivery of insulin to the brain, and low binding affinity of insulin for its receptors, lead to a state of insulin resistance in the brain. Interestingly, this physiological decrease in insulin signaling seems more marked in neurodegenerative diseases such as AD[61].
ROLE OF lNSULlN RESlSTANCE lN AD
Several studies have reported that peripheral insulin resistance can promote the onset of AD by reducing insulin supply to the brain and increasing levels of amyloid beta (Aβ), phosphorylation of tau protein, oxidative stress; pro-inflammatory cytokines, production of advanced glycation products(AGEs), advanced glycation end products; dyslipidemia, and apoptosis[62]. However, it has been proposed that insulin resistance is not limited to peripheral tissues, and, in particular, that the brain itself could become insulin resistant with or without the presence of DM-2, and that this could promote(or even lead to) the appearance of key pathophysiological factors of the disease[11,60]. Some researchers have even used the term "type 3 diabetes" to explain these phenomena[60]. But this new definition is questioned by other authors who prefer the term "insulin resistant brain state"[11,63].
Likewise, several insulin signaling markers have been found in the brain of AD patients[60,63], and the selective increase in brain insulin of patients with AD or high risk of AD, by intranasal insulin administration, resulted in improved memory functions[64].
When examining the existence of brain insulin resistance in patients with AD and mild cognitive disorder, it has been shown that the hippocampus, and to a lesser extent the cerebellar cortex, of patients with AD show a reduction in the insulin signaling pathway by IR -> IRS-1 -> PI3K -> AKT and IGF-1 by IGF-1R -> IRS -2 -> PI3K, compared to healthy tissue. This dysfunction occurred independently of diabetic status and APOE ε4 genotype, and gradually worsened as the AD progressed;furthermore, there was a reduced activation of this same pathway when comparing patients with established AD with healthy controls, despite increasing the insulin dose tenfold[11]. These results were associated with elevated levels of phosphorylated IRS-1 proteins at serine residues 636 and 616 (IRS-1 pSer) (which inhibit insulin signaling). Other studies have also shown elevated levels of the IRS-1 proteins pSer312 and pSer616 in association with neuronal insulin resistance in AD[63], leading some authors to suggest that the detection of elevated protein levels of IRS-1 phosphorylated on serine residues could serve as a potential biomarker of neuronal insulin resistance in AD, as is already the case for insulin resistance in peripheral tissues.
The participation of brain insulin resistance in AD has been demonstrated but the obligatory questions are: Is the observed insulin resistance due to inherent resistance of IRs, or to alterations in the transit of insulin across the BBB? Is the involvement of insulin resistance in AD primary (isolated manifestation with selective brain damage) or secondary (after systemic insulin resistance due to obesity, DM, nonalcoholic fatty liver disease, or metabolic syndrome)?
Who knows for what good end he may have done this thing? So they went on their way and entered the palace, and there in the hall stood a cupboard in which lay the ready-made bridal shirt, looking for all the world as though it were made of gold and silver
The effects of insulin result from its binding to a specific membrane receptor, the IR, and are expressed primarily in its three target tissues: liver, muscle, and adipose tissue. However, this receptor is also present on nerve endings in key brain regions such as the olfactory bulb, hypothalamus, cerebral cortex; cerebellum, and hippocampus. It should be noted that there is a striking similarity between the IR and the IGF-1 receptor (IGF-1R) in various brain regions, which could lead to an overlap of signal transduction pathways and to the same neuronal effects[56,57].
And he stared hard at the princess as she clapped her hands with joy and ran up to them, crying, Oh, do let us keep that delicious beast for to-night; it will make such a nice plaything
The role of insulin in modulating cognition is suggested in part by the high density of IRs in the hippocampus, cortex, and amygdala, and by the fact that these receptors increase in response to spatial memory training[72]. Furthermore, the acute administration of insulin improves memory performance in rats, and improves verbal memory, attention, and cognition in humans, by activating IRs in the hippocampus. Nevertheless, there is conflicting evidence from studies using NIRKO mice (transgenic mice in which the IR gene has been deleted in the brain). NIRKO mice do not show alterations in memory performance, suggesting that other mechanisms may also contribute[72]. However, NIRKO mice have increased glycogen synthase kinase 3 beta (GSK3-beta) activity, resulting in hyperphosphorylation of tau protein[72]. Conversely, a growing body of evidence indicates that insulin plays an important role in cognition, and although the underlying molecular mechanisms remain unclear,studies indicate that insulin resistance in the hippocampus is a risk factor for cognitive decline and dementia in AD[73].
PROTElN AGGREGATlON lN AD: B-AMYLOlD PEPTlDE AND TAU PROTElN
β-Amyloid peptide
The term β-amyloid peptide refers to a collection of peptides of 39 to 43 amino acids in length that are formed by cleavage of the amyloid precursor protein (APP) under the action of β and γ secretases. They are products of normal cellular metabolism and probably have a physiological role that is not yet fully understood. The abnormal oligomerization of some of these peptides (such as Aβ-42) and the formation of extracellular plaques containing Aβ fibrils (neuritic plaques) in their center, constitute one of the histopathological markers of AD in post-mortem brain tissue. In sporadic AD, part of the oligomerization of Aβ may be caused by decreased degradation and elimination of Aβ from the brain. The Aβ can be degraded by various peptidases such as IDE, neprisilin, angiotensin-converting enzyme, and many serine proteases. Recent
studies have shown that insulin resistance can contribute to amyloid deposits in the frontal and temporal areas in asymptomatic subjects[50].
Experimentally, the induction of insulin resistance in rats increases the production of Aβ by increasing the activation of β-secretase and γ-secretase, and by decreasing the levels of the IDE[46].Insulin and IGF-1, by activating the PI3K/MAPK pathways, stimulate the transport and clearance of Aβ out of the central nervous system by increasing the expression of Aβ transporters in cerebrospinal fluid.On the other hand, the accumulation of Aβ could also be explained by the effect of insulin on IDE. The main function of IDE is to break down insulin; however, Aβ is also a substrate for IDE, but with lower affinity. Therefore, when insulin rises, it inhibits the breakdown of Aβ by IDE; nevertheless, Aβ can be degraded by others[65].
The activity of IDE decreases with age and there is a reduction in its activity similarly in patients and experimental models of AD[65]. Thus, insulin resistance through decreased IDE activity or, to a lesser extent, competitive inhibition of IDE by insulin during prolonged hyperinsulinism, could lead to defects of IDE autophagy, and, consequently, to a decrease in the turnover and/or neutralization of amyloidogenic proteins in β cells, as well as a defect in the degradation of Aβ that would promote AD-related neuropathological lesions[65]. Therefore, insulin resistance is believed to play a role in the accumulation of Aß, one of the key markers of the pathogenesis of DM-2.
Tau protein
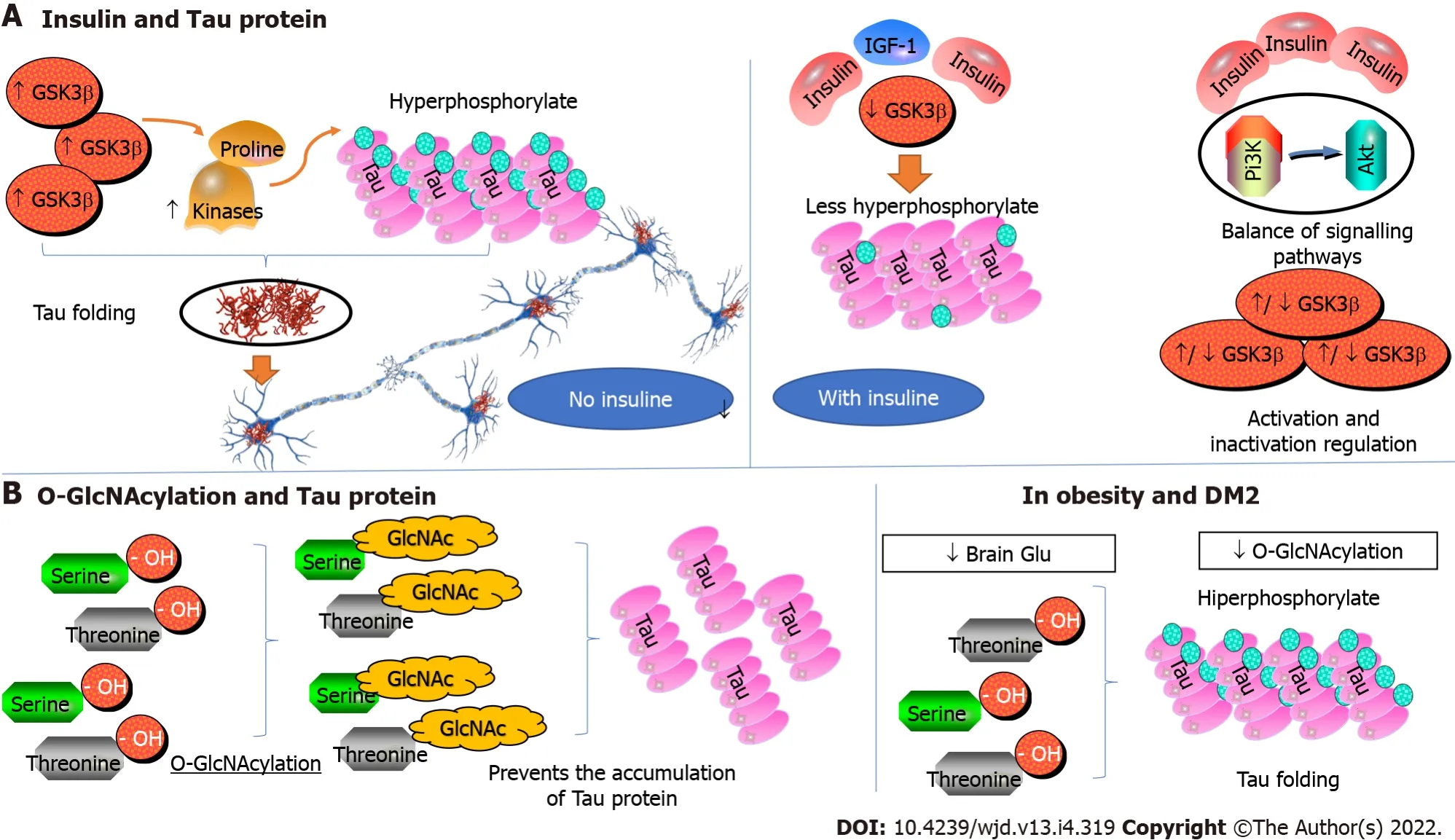
Tau is a protein that belongs to MAPs (microtubule-associated proteins). In its primary conformation,the tau protein is a soluble and unfolded protein that participates in the stabilization of microtubules and in the axonal growth of neurons. In AD, however, tau is hyperphosphorylated due to inappropriate activation of several proline-directed kinases, including glycogen synthase kinase-3 beta (GSK3β). This results in the folding of the tau protein and self-aggregation into insoluble fibrillar fiber structures(paired helical filaments and straight filaments) that form neurofibrillary tangles, dystrophic neurites,and neuropil threads[66].
Insulin regulates GSK3β; therefore, insulin and IGF-1, by inhibiting GSK3β, inhibit the phosphorylation of tau, and enhance its binding to microtubules. Insulin manages the balance of tau phosphorylation through subsequent activation and inactivation of GSK3β through the Pi3K/Akt signaling pathway[66]. The GSK3β is one of the major signaling molecules downstream of Akt, and the insulin signaling defect in obesity and DM-2 results in aberrant activation of GSK3β leading to increased phosphorylation and accumulation of tau. Therefore, the molecular pathways of DM and AD run parallel in the pathogenesis of these diseases, and the Pi3K/Akt signaling pathway plays a critical role in the neuropathology of tau. Likewise, the alteration of glucose metabolism due to insulin resistance can affect the pathology of tau through the dysregulation of O-linked-N-acetylglucosaminylation (OGlcNAcylation)[66].
As I read this message, a wave of sadness ran through me and I realized that she must have thought she was writing to her father the whole time. She and I would never have openly exchanged such words of affection. Feeling guilty for not clarifying, yet not wanting to embarrass her, I simply responded, Love you too! Have a good sleep!
Studies of the brains of patients who died of AD and had vascular dementia showed that the signs of both diseases were often found simultaneously. The researchers concluded that both causes could contribute to the clinically observed difficulties, which leads us to then speak of mixed dementia (AD +vascular cognitive decline)[26] (Table 2).
Like phosphorylation, O-GlcNAcylation is a dynamic post-translational modification that involves the attachment of N-acetyl-d-glucosamine (GlcNAc) residues to the hydroxyl group of serine and threonine residues, and it is deregulated in obesity and DM-2. Decreased brain glucose metabolism and O-GlcNAcylation have been shown to lead to hyperphosphorylation of tau in both
and
models. By contrast, increased O-GlcNAcylation prevents the pathological accumulation of tau.Moreover, in experimental models, the specific elimination of insulin in the neurons of NIRKO(Neuronal Insulin Receptor Knockout) mice leads to hyperphosphorylation of tau associated with a decrease in phosphorylation of Akt and GSK3β. Similarly, the insulin signaling defect in IRS-2 knockout(KO) mice results in the accumulation of hyperphosphorylated tau protein. This accumulation has been attributed to protein phosphatase 2a (PP2A), an enzyme responsible for the dephosphorylation of tau.However, a decrease in GSK3β dephosphorylation was found in this same animal model, suggesting that GSK3β, again, could be responsible for the accumulation of hyperphosphorylated tau[67].
The patterns of tau phosphorylation between NIRKO mice and IRS-2 KO mice vary, suggesting that tau phosphorylation could be controlled not only by insulin resistance but also by other factors such as hyperinsulinism, hyperglycemia, and inflammation. Increased insoluble hyperphosphorylated tau protein and deposition of neurofibrillary tangles occur in various animal models of obesity, DM-2, or AD, with altered insulin signaling. Therefore, altered insulin signaling could promote the formation of neurofibrillary degeneration, disrupt neural cytoskeletal networks and axonal transport, and lead to loss of synaptic connections and progressive neurodegeneration[68] (Figure 3).
Vascular dementia and AD are two types of dementia. They share similarities but differ in terms of causes, the course of the disease, and the type of impairment of cognitive abilities, among other things.The cause of AD is still unknown. However, it is known to be associated with the death of neurons in certain areas of the brain. The affected areas present characteristic abnormalities (neuritic plaques and neurofibrillary tangles)[24,25]. In general, the areas involved in memory are affected first, then the damage gradually spreads to other areas of the brain[25]. In vascular dementia, it is the cerebral vascular network that is involved and there may be a multi-infarct vascular dementia (2 or more simultaneous or asynchronous arterial or venous territorial ischemic vascular events), a vascular dementia as a sequel to a heart attack or hemorrhage in some strategic topography, or a mild or greater cognitive impairment associated with confluent and progressive subcortical lesions of the cerebral microcirculation. This vascular network refers to everything that has to do with blood vessels including veins, arteries, and capillaries. Since the vessels that can be affected are numerous, the clinical picture is more variable than in AD. Here, it is not necessarily the areas responsible for memory that are initially affected[22].
The combination of all the aforementioned elements described, demonstrate the effects that the insulin/IGF-1 signaling defect could have on tau aggregation in AD.
BRAlN GLUCOSE METABOLlSM
There may also be a relevant relationship between neurodegeneration, insulin signaling, and glucose utilization in the brain. Glucose metabolism and insulin signaling are essential for normal brain function, and circulating glucose levels play an important role in learning and memory functions.
They came to her when they were quite tiny, and never left her until they were grown up and had to go away into the great world; and when that time came she gave to each whatever gift he asked of her
Brain glucose metabolism in AD
One of the pathological features of AD is the extreme drop in energy metabolism in the affected areas of the brain. Regional patterns of brain hypometabolism are seen in the early stage of AD. Glucose is necessary for the synthesis of several neurotransmitters such as acetylcholine, dopamine, γaminobutyric acid (GABA), and glutamate,
, which mainly participate in synaptic plasticity and cognitive functions[70]. However, cerebral glucose hypometabolism is not directly related to insulin resistance in the brain since, unlike in the periphery, this resistance does not affect neuronal glucose uptake[11]. Rather, glucose hypometabolism in AD may be the result of reduced postsynaptic neurotransmission (a likely effect of reduced insulin signaling in the brain). This is because glutamate and other depolarizing agents stimulate glucose uptake in the brain and the potency of this effect is reduced in AD[11].
NEUROlNFLAMMATlON lN AD
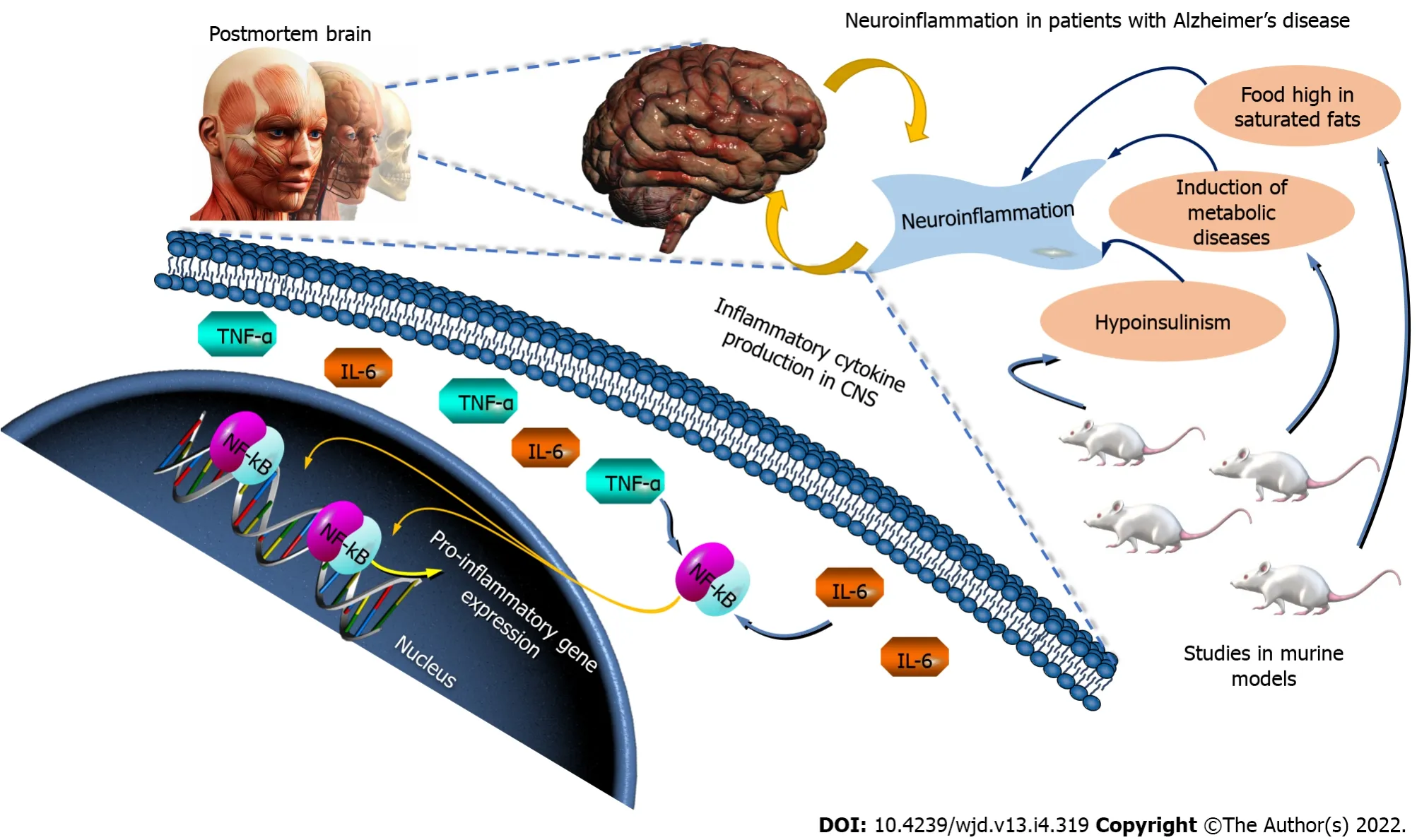
Numerous studies have shown the presence of inflammatory markers in the post-mortem brains and blood of AD patients. There is considerable evidence to suggest that inflammation modulates cognition;from post mortem studies, to analyses of genomic association, to
models addressing its use (for example, of chronic injections of lipopolysaccharide (used to induce inflammation) that accelerates the progression of AD), or from human studies showing that patients with systemic infection have greater cognitive impairment. The induction of metabolic diseases in rodents also induces neuroinflammation:mice fed a high-fat diet showed a higher expression of inflammatory markers in their brains, and cognitive impairment[71]. Furthermore, hyperinsulinism associated with insulin resistance leads to the production of cytokines in the central nervous system[71]. The tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), which are pro-inflammatory cytokines, are known to activate the nuclear factor kappa B (NF-κB) pathway that leads to the transcription of pro-inflammatory genes that exacerbate this cycle. Thus, chronic inflammation may represent an underlying mechanism common to AD and metabolic disorders (Figure 4).
SYNAPTlC TRANSMlSSlON AND MEMORY
Tsar Dolmat was greatly angered, and shouted in a loud voice: How now! This is a fine, bold handed Cossack to be caught in such a shameful28 theft! Who art thou, from what country comest thou? Of what father art thou son, and how art thou named?
Two pathways are involved in intracellular events after the binding of insulin and IGF-1 at its receptor and the activation of its tyrosine kinase function of its intracellular domain: the Shc proteins(Src homologous and collagen protein ) that activate the MAP kinase (mitogen-activated protein)pathway, resulting in the translocation of extracellular signal-regulated protein kinases (ERK) to the nucleus and the effects of mitogenic insulin; and, on the other hand, the IRS proteins (IR substrate 1 and 2), which activate the phosphatidylinositol-3 kinase (PI3K)/AKT (also called protein kinase B) pathway upon which the metabolic effects depend. Conversely, the phosphorylation of IRS proteins on serine residues by protein kinases c-JUN kinases (JNK), atypical protein kinases C (PKC), and inhibitor of nuclear factor kβ kinase (IKKβ), plays an antagonistic role to that of the phosphorylation of only tyrosine residues, and causes the dissociation of IRS proteins from the IR or the IGF-1 receptor, and promotes their degradation; thus inhibiting the downstream insulin/IGF-1 signaling. This suggests that in maintaining the protein stability, IRS acts as a critical point in the insulin signaling pathway, and can determine the extent of its actions. In addition, negative control of the insulin signal may come from the degradation of the hormone by the insulin-degrading enzyme (IDE), or by dephosphorylation of the receptor or IRS proteins by tyrosine phosphatases PTPases such as PTP1B and LAR (a molecule related to the common antigen of leukocytes)[57,58].
lNSULlN RESlSTANCE: CAUSE OR CONSEQUENCE OF AD?
A study in a population with AD found that up to 80% of patients had DM-2 or insulin resistance,suggesting that AD may lead to the manifestation of DM. Unfortunately, there are no longitudinal studies that demonstrate whether the presentation of DM occurs after the onset of AD or if it precedes the AD diagnosis. Tissue analysis confirms that insulin resistance in the brain deteriorates with the progression of AD, but when it appears in the progression of the disease is unknown. Additionally,studies have shown that Aβ can also affect insulin signaling in multiple ways[52]: Competing with or reducing the affinity of insulin for its own receptor; Inhibiting IR autophosphorylation and subsequent activation of PI3K/Akt; Causing inhibitory phosphorylation on a serine residue of IRS by stimulation of the N-terminal kinase c-Jun (JNK); and Inhibiting the activation of Akt preventing its interaction with isoenzyme 1 of the pyruvate dehydrogenase-lipoamide kinase (PDK1).
The Aβ derivatives (ADDLs) induce abnormal IR expression and disrupt insulin signaling, thus potentially contributing to the development of central insulin resistance. There is also the question of the involvement of tau protein in insulin resistance, as researchers very recently demonstrated that mice deficient in tau protein develop an abnormal response to insulin in the hippocampus. The question now is whether an abnormal tau protein (such as the hyperphosphorylated tau protein in AD) could have the same impact on brain insulin resistance[75].
If the faulty insulin/IGF1 pathway leads to inflammation, it has also been proposed that neuroinflammation in AD may lead to faulty insulin signaling in neurons. For example, inflammation associated with peripheral infections produces a peripheral inflammatory response that, in turn, can reach the brain without knowing by what process.
At the molecular and cellular level, when cytokine receptors (such as TNF-α) are activated, cellular stress pathways (JNK and IKK) that cause phosphorylation at a serine residue of IRS-1 are activated,and thus inhibit intracellular signaling of insulin in the brain. The TNF-α is secreted in the brain in response to infections and abnormal protein aggregation, and is increased in the cerebrospinal fluid of AD patients and transgenic mouse models. Initial evidence linking inflammatory pathways and the disruption of insulin/IGF1 signaling in AD, comes from the observation that Aβ induces IRS-1 inhibition through the TNF-α[75] and the removal of IRs from cell membranes. Based on this evidence, it seems plausible to postulate that inflammation plays a role in disrupting insulin/IGF1 signaling pathways in AD and metabolic disorders[76] (Figure 6).

AMYLlN AS A LlNK BETWEEN DM-2 AND NEURODEGENERATlON: AD
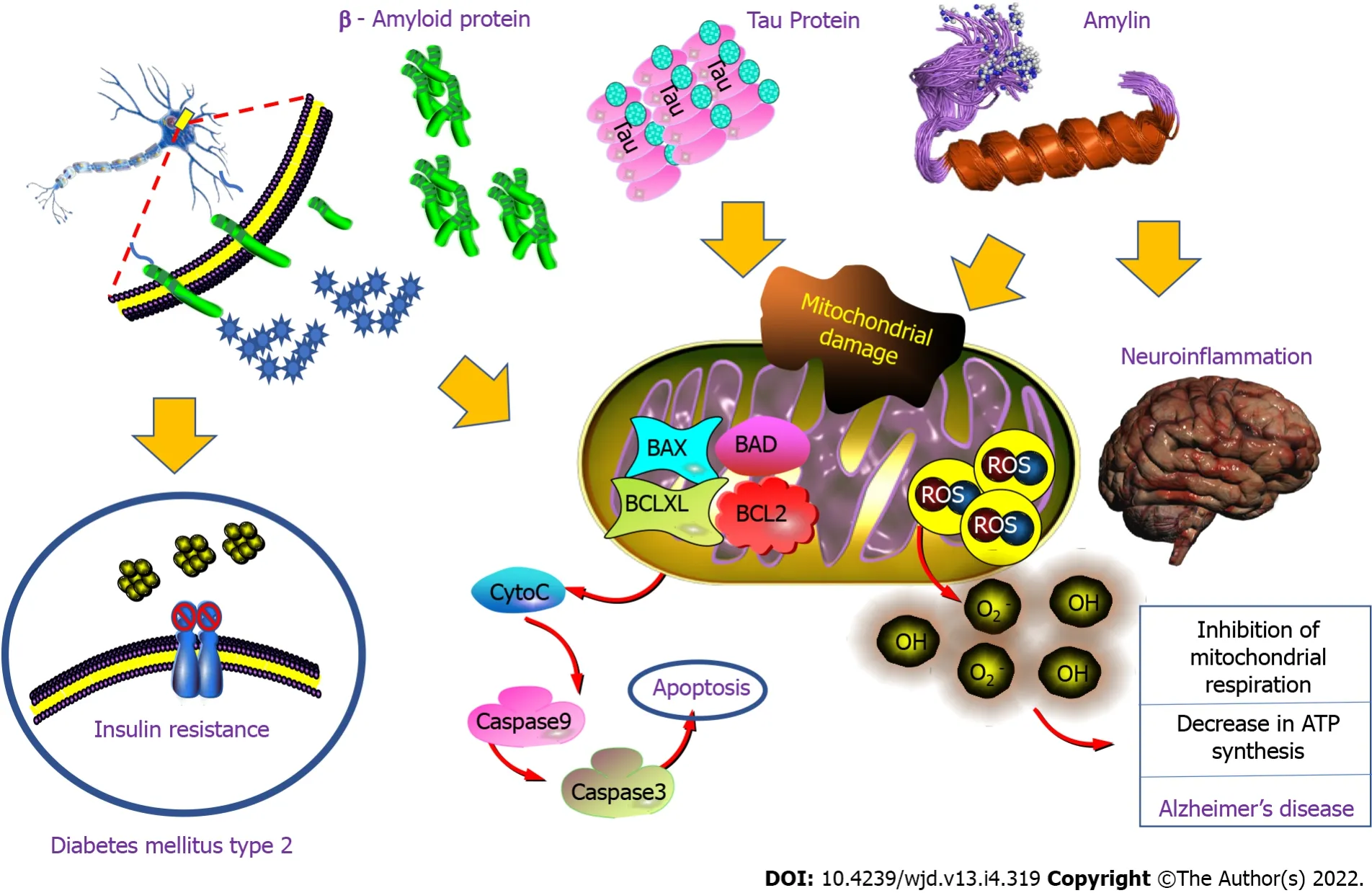
Amylin polypeptide is a pancreatic hormone that is elevated as a crucial component in the etiology of DM-2 and AD[77]. Recent studies have shown that people with DM-2 are twice as likely as the rest of the population to suffer from AD, which is the leading cause of dementia worldwide. Therefore, there must be a biological process that facilitates the development of neurodegeneration in insulin resistant patients. These two pathologies, as we know, have some points in common: Both are particularly favored by obesity; They are also associated with groups of proteins from the same family (that of amyloids); In the case of DM, it is amylin (an endogenous protein with a hyperglycemic effect) that accumulates in the pancreas; In AD, it is β-amyloids that accumulate in plaques between neurons.
However, these two peptides have been found in the pancreas of people with DM and their presence seems to coincide with the progression of the disease. Amylin is a hormonal peptide released by pancreatic beta cells just like insulin. The coexistence of β-amyloid, tau protein, and amylin in the brain and pancreas, demonstrates their ability to promote amyloid accumulation and cell dysfunction,neuronal dysfunction, and pancreatic β-amyloid accumulation. That is, β-amyloid protein, tau protein,and amylin can interact synergistically, increasing to promote amyloid deposition, oxidative stress,mitochondrial dysfunction; inflammation, insulin resistance, and cell damage, culminating in dysregulation of amyloid metabolism, and glucose and neurodegeneration in AD and DM-2. In previous studies, it has been found that high plasma amylin concentration is associated with the incidence of AD,which suggests that amylin is a risk factor for AD[78,79]; and, amylin deposits in the brain have been found to interact with the β-amyloid protein and with tau protein in the pancreas and the hippocampus,which provides new evidence of a potential overlap between the understanding of the mechanisms of the pathophysiology of DM-2 and AD[80] (Figure 7).
MlTOCHONDRlAL DYSFUNCTlON AND OXlDATlVE STRESS lN AD
Metabolic dysfunction derived from oxidative stress and altered mitochondrial function has been extensively documented in AD. For example, increased levels of oxidative stress markers are correlated with the diminution in mitochondrial enzymatic activity in several brain areas and in peripheral tissues from AD patients[81,82]. The brain is very susceptible to oxidative stress due to its high content of polyunsaturated fatty acids, high oxygen comsumption, and low scavenging antioxidant capacity response system. Mitochondria are both generators and direct targets of reactive oxygen and reactive nitrogen species, thus oxidative stress and decreased mitochondrial activity of the oxidative phosphorylation system could have a significant effect on neuronal integrity.
Yes. I mean, no. He just doesn t holler when he sees a bug crawling out of a peach seed or anything. He just looks at it carefully. But it is just a bug, isn t it, really?
Additionally, aging, a common risk factor for AD and insulin resistance, is also associated with decreased antioxidant capacity, increased oxidative stress, and decreased mitochondrial function.
The production of low levels of reactive oxygen species is not always detrimental: in peripheral tissues, the transient generation of reactive oxygen species facilitates insulin/IGF1 signaling by inhibiting phosphatases such as PTEN (which normally invert the PI3K signal)[82]. In the brain, the transient production of reactive oxygen species is used for synaptic transmission, facilitating long-term potentiation[82,83].
The first evidence for a link between insulin/IGF-1 signaling and mitochondrial dysfunction comes from experiments showing that insulin prevented the depolarization of the inner mitochondrial membrane in sensory neurons of diabetic rats[84]. Consistent with this, the activity of ATPase and the levels of the mitochondrial coenzyme Q9 were reduced in a model of DM. Thus, a decrease in the antioxidant activity of the mitochondria due to aging or alteration of insulin/IGF-1 signaling, in the case of metabolic disorders, could increase the vulnerability to AD since insulin prevents the decrease of oxidative phosphorylation and reduces oxidative stress induced by Aβ. Insulin stimulates mitochondrial protein synthesis, IGF-1 protects against hyperglycemia-induced oxidative stress, and the insulin/IGF-1 signaling defects makes neurons more vulnerable to reactive oxygen species[85]. Additionally, insulin regulates mitochondrial dynamics, autophagia, and tau phosphorylation in diabetic rats[86], and abolishes the Ab-40 mediated mitochondrial alterations[85], and increases mitochondrial ATP production in neuronal cells[87].
Seeing all this, the four dragons felt very sorry, for they knew the Jade Emperor only cared about pleasure, and never took the people to heart. They could only rely on themselves to relieve the people of their miseries. But how to do it?
Regardless of the exact sequence of events leading to neurodegeneration, there is strong evidence that mitochondrial dysfunction plays a key role in animal models of AD and DM. For example, the triple transgenic mouse model of AD (that expresses mutants of human APP, presenilin 1, and tau protein)presents both neuritic plaques and neurofibrillary tangles in brain. Furthermore, that triple transgenic model showed diminished mitochondrial respiratory activity and impaired oxidative phosphorylation systems. These alterations are correlated with the loss of synaptic integrity, with increased levels of reactive oxygen species, and with decreased levels of reduced glutathione and vitamin E. Interestingly,that alterations are comparable to those induced in wild-type mice treated with sucrose, is consistent with the proposal that mitochondrial alterations are associated with DM and that they contribute to the development of AD[88]. In accordance, NIRKO mice showed mitochondrial dysfunction, altered mitochondrial morphology, reduced expression of proteins of the oxidative phosphorylation system,and diminished mitochondrial respiratory activity(March 17,2022). Hence, alterations in the insulin/IGF-1 signaling, in DM for example, could lead to mitochondrial dysfunction, which would reduce the antioxidant capacity of the cell, and, when aggravated by aging, this would increase the vulnerability of the cell to Aβ-induced toxicity.
CONCLUSlON
Finally, the appearance of cognitive impairement in DM-2 contributes to making the patient even more fragile and increasingly necessitating continuous home care to perform daily activities, which impacts the patients’ quality of life. Interestingly, the insulin signaling pathway is hypothesized to be a specific target of therapeutic strategies to decrease alterations associated with age-related cognitive decline.
FOOTNOTES
Ortiz GG contributed coordination and work writing; Huerta M contributed diabetes expert and reviewer, and amyline contribution; González-Usigli HA and Mireles-Ramírez MA contributed neurologists and clinical work reviewers and writing; Torres-Sánchez ED and Delgado-Lara DL updated analysis of references, and graphic art; Pacheco-Moisés FP, Torres-Mendoza BM and Moreno-Cih RI contributed basic concepts, and their writing; Velázquez-Brizuela IE and Ortiz GG contributed neuropsychology and dementia concepts and writing.
This work does not present any conflict of interest.
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Mexico
Genaro G Ortiz 0000-0002-7054-3313; Miguel Huerta 0000-0001-8515-0777; Héctor A González-Usigli 0000-0002-3906-9051; Erandis D Torres-Sánchez 0000-0002-8131-6853; Daniela LC Delgado-Lara 0000-0002-1968-4815; Fermín P Pacheco-Moisés 0000-0002-3769-5649; Mario A Mireles-Ramírez 0000-0002-8321-7883; Blanca MG Torres-Mendoza 0000-0003-2233-571X; Roxana I Moreno-Cih 0000-0002-9294-3318; Irma E Velázquez-Brizuela 0000-0002-3050-9575.
Gao CC
A
Wu RR
1 Chawla A, Chawla R, Jaggi S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum?
2016; 20: 546-551 [PMID: 27366724 DOI: 10.4103/2230-8210.183480]
2 Corcóstegui B, Durán S, González-Albarrán MO, Hernández C, Ruiz-Moreno JM, Salvador J, Udaondo P, Simó R. Update on Diagnosis and Treatment of Diabetic Retinopathy: A Consensus Guideline of the Working Group of Ocular Health(Spanish Society of Diabetes and Spanish Vitreous and Retina Society).
2017; 2017: 8234186 [PMID:28695003 DOI: 10.1155/2017/8234186]
3 Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities.
2017; 12: 2032-2045 [PMID: 28522654 DOI: 10.2215/CJN.11491116]
4 Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, Freeman R, Truini A, Attal N, Finnerup NB,Eccleston C, Kalso E, Bennett DL, Dworkin RH, Raja SN. Neuropathic pain.
2017; 3: 17002 [PMID:28205574 DOI: 10.1038/nrdp.2017.2]
5 Weledji EP, Fokam P. Treatment of the diabetic foot - to amputate or not?
2014; 14: 83 [PMID: 25344293 DOI:10.1186/1471-2482-14-83]
6 Chornenkyy Y, Wang WX, Wei A, Nelson PT. Alzheimer's disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline.
2019; 29: 3-17 [PMID:30106209 DOI: 10.1111/bpa.12655]
7 Saedi E, Gheini MR, Faiz F, Arami MA. Diabetes mellitus and cognitive impairments.
2016; 7: 412-422[PMID: 27660698 DOI: 10.4239/wjd.v7.i17.412]
8 Williamson JD, Miller ME, Bryan RN, Lazar RM, Coker LH, Johnson J, Cukierman T, Horowitz KR, Murray A, Launer LJ; ACCORD Study Group. The Action to Control Cardiovascular Risk in Diabetes Memory in Diabetes Study(ACCORD-MIND): rationale, design, and methods.
2007; 99: 112i-122i [PMID: 17599421 DOI:10.1016/j.amjcard.2007.03.029]
9 Sherwani SI, Khan HA, Ekhzaimy A, Masood A, Sakharkar MK. Significance of HbA1c Test in Diagnosis and Prognosis of Diabetic Patients.
2016; 11: 95-104 [PMID: 27398023 DOI: 10.4137/BMI.S38440]
10 Jayaraman A, Pike CJ. Alzheimer's disease and type 2 diabetes: multiple mechanisms contribute to interactions.
2014; 14: 476 [PMID: 24526623 DOI: 10.1007/s11892-014-0476-2]
11 Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS,Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline.
2012; 122: 1316-1338 [PMID: 22476197 DOI: 10.1172/JCI59903]
12 Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP Jr, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus.
2009; 301: 1565-1572 [PMID: 19366776 DOI: 10.1001/jama.2009.460]
13 Kennelly SP, Lawlor BA, Kenny RA. Blood pressure and dementia - a comprehensive review.
2009; 2: 241-260 [PMID: 21179532 DOI: 10.1177/1756285609103483]
14 Trompet S, van Vliet P, de Craen AJ, Jolles J, Buckley BM, Murphy MB, Ford I, Macfarlane PW, Sattar N, Packard CJ,Stott DJ, Shepherd J, Bollen EL, Blauw GJ, Jukema JW, Westendorp RG. Pravastatin and cognitive function in the elderly.Results of the PROSPER study.
2010; 257: 85-90 [PMID: 19653027 DOI: 10.1007/s00415-009-5271-7]
15 Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D,Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S; American Heart Association Stroke Council, Council on Epidemiology and Prevention,Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association.
2011; 42: 2672-2713 [PMID:21778438 DOI: 10.1161/STR.0b013e3182299496]
16 Albai O, Frandes M, Timar R, Roman D, Timar B. Risk factors for developing dementia in type 2 diabetes mellitus patients with mild cognitive impairment.
2019; 15: 167-175 [PMID: 30655669 DOI:10.2147/NDT.S189905]
17 Li X, Song D, Leng SX. Link between type 2 diabetes and Alzheimer's disease: from epidemiology to mechanism and treatment.
2015; 10: 549-560 [PMID: 25792818 DOI: 10.2147/CIA.S74042]
18 Ryan JP, Fine DF, Rosano C. Type 2 diabetes and cognitive impairment: contributions from neuroimaging.
2014; 27: 47-55 [PMID: 24394151 DOI: 10.1177/0891988713516543]
19 Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review.
2006; 5: 64-74 [PMID: 16361024 DOI: 10.1016/S1474-4422(05)70284-2]
20 Cioffi F, Adam RHI, Broersen K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer's Disease.
2019; 72: 981-1017 [PMID: 31744008 DOI: 10.3233/JAD-190863]
21 Santos CY, Snyder PJ, Wu WC, Zhang M, Echeverria A, Alber J. Pathophysiologic relationship between Alzheimer's disease, cerebrovascular disease, and cardiovascular risk: A review and synthesis.
2017; 7: 69-87 [PMID: 28275702 DOI: 10.1016/j.dadm.2017.01.005]
22 Iadecola C. The pathobiology of vascular dementia.
2013; 80: 844-866 [PMID: 24267647 DOI:10.1016/j.neuron.2013.10.008]
23 Hugo J, Ganguli M. Dementia and cognitive impairment: epidemiology, diagnosis, and treatment.
2014;30: 421-442 [PMID: 25037289 DOI: 10.1016/j.cger.2014.04.001]
24 Kumar A, Sidhu J, Goyal A, Tsao JW. Alzheimer Disease. 2021 Aug 11. In: StatPearls [Internet]. Treasure Island (FL):StatPearls Publishing; 2022 Jan- [PMID: 29763097]
25 Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century.
2011; 3:77sr1 [PMID: 21471435 DOI: 10.1126/scitranslmed.3002369]
26 Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer's disease--lessons from pathology.
2014; 12: 206 [PMID: 25385447 DOI: 10.1186/s12916-014-0206-2]
27 Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia.
2010; 68 Suppl 2: S74-S87 [PMID: 21091952 DOI: 10.1111/j.1753-4887.2010.00352.x]
28 Cade WT. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting.
2008; 88:1322-1335 [PMID: 18801863 DOI: 10.2522/ptj.20080008]
29 Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer's disease.
2016; 131: 659-685 [PMID: 27062261 DOI: 10.1007/s00401-016-1571-z]
30 Rabinovici GD, Stephens ML, Possin KL. Executive dysfunction.
2015; 21: 646-659 [PMID:26039846 DOI: 10.1212/01.CON.0000466658.05156.54]
31 Koekkoek PS, Kappelle LJ, van den Berg E, Rutten GE, Biessels GJ. Cognitive function in patients with diabetes mellitus:guidance for daily care.
2015; 14: 329-340 [PMID: 25728442 DOI: 10.1016/S1474-4422(14)70249-2]
32 Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes--systematic overview of prospective observational studies.
2005; 48: 2460-2469 [PMID: 16283246 DOI: 10.1007/s00125-005-0023-4]
33 Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study.
1999; 53: 1937-1942 [PMID: 10599761 DOI: 10.1212/wnl.53.9.1937]
34 Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease.
2005; 65: 545-551 [PMID: 16116114 DOI: 10.1212/01.wnl.0000172914.08967.dc]
35 Gudala K, Bansal D, Schifano F, Bhansali A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies.
2013; 4: 640-650 [PMID: 24843720 DOI: 10.1111/jdi.12087]
36 Wang KC, Woung LC, Tsai MT, Liu CC, Su YH, Li CY. Risk of Alzheimer's disease in relation to diabetes: a populationbased cohort study.
2012; 38: 237-244 [PMID: 22572745 DOI: 10.1159/000337428]
37 Huang CC, Chung CM, Leu HB, Lin LY, Chiu CC, Hsu CY, Chiang CH, Huang PH, Chen TJ, Lin SJ, Chen JW, Chan WL. Diabetes mellitus and the risk of Alzheimer's disease: a nationwide population-based study.
2014; 9:e87095 [PMID: 24489845 DOI: 10.1371/journal.pone.0087095]
38 Xu WL, von Strauss E, Qiu CX, Winblad B, Fratiglioni L. Uncontrolled diabetes increases the risk of Alzheimer's disease:a population-based cohort study.
2009; 52: 1031-1039 [PMID: 19280172 DOI: 10.1007/s00125-009-1323-x]
39 Tuligenga RH, Dugravot A, Tabák AG, Elbaz A, Brunner EJ, Kivimäki M, Singh-Manoux A. Midlife type 2 diabetes and poor glycaemic control as risk factors for cognitive decline in early old age: a post-hoc analysis of the Whitehall II cohort study.
2014; 2: 228-235 [PMID: 24622753 DOI: 10.1016/S2213-8587(13)70192-X]
40 Tolppanen AM, Lavikainen P, Solomon A, Kivipelto M, Uusitupa M, Soininen H, Hartikainen S. History of medically treated diabetes and risk of Alzheimer disease in a nationwide case-control study.
2013; 36: 2015-2019[PMID: 23340883 DOI: 10.2337/dc12-1287]
41 Cheng G, Huang C, Deng H, Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a metaanalysis of longitudinal studies.
2012; 42: 484-491 [PMID: 22372522 DOI:10.1111/j.1445-5994.2012.02758.x]
42 Zhang J, Chen C, Hua S, Liao H, Wang M, Xiong Y, Cao F. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer's disease.
2017; 124: 41-47 [PMID: 28088029 DOI: 10.1016/j.diabres.2016.10.024]
43 Kim B, Feldman EL. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome.
2015; 47: e149 [PMID: 25766618 DOI: 10.1038/emm.2015.3]
44 S Roriz-Filho J, Sá-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, Moriguti JC, Roriz-Cruz M.(Pre)diabetes, brain aging, and cognition.
2009; 1792: 432-443 [PMID: 19135149 DOI:10.1016/j.bbadis.2008.12.003]
45 Frisardi V, Solfrizzi V, Seripa D, Capurso C, Santamato A, Sancarlo D, Vendemiale G, Pilotto A, Panza F. Metaboliccognitive syndrome: a cross-talk between metabolic syndrome and Alzheimer's disease.
2010; 9: 399-417[PMID: 20444434 DOI: 10.1016/j.arr.2010.04.007]
46 Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM.Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease.
2004;18: 902-904 [PMID: 15033922 DOI: 10.1096/fj.03-0978fje]
47 de la Monte SM, Longato L, Tong M, Wands JR. Insulin resistance and neurodegeneration: roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis.
2009; 10: 1049-1060 [PMID: 19777393]
48 Stanley M, Macauley SL, Holtzman DM. Changes in insulin and insulin signaling in Alzheimer's disease: cause or consequence?
2016; 213: 1375-1385 [PMID: 27432942 DOI: 10.1084/jem.20160493]
49 de la Monte SM. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer's Disease.
2017; 77: 47-65 [PMID: 27988872 DOI: 10.1007/s40265-016-0674-0]
50 Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, Craft S, Oh J, Statz E, Hermann BP, Jonaitis EM, Koscik RL, La Rue A, Asthana S, Bendlin BB. Insulin resistance predicts brain amyloid deposition in late middle-aged adults.
2015; 11: 504-510.e1 [PMID: 25043908 DOI: 10.1016/j.jalz.2014.03.011]
51 Ribe EM, Lovestone S. Insulin signalling in Alzheimer's disease and diabetes: from epidemiology to molecular links.
2016; 280: 430-442 [PMID: 27739227 DOI: 10.1111/joim.12534]
52 Rani V, Deshmukh R, Jaswal P, Kumar P, Bariwal J. Alzheimer's disease: Is this a brain specific diabetic condition?
2016; 164: 259-267 [PMID: 27235734 DOI: 10.1016/j.physbeh.2016.05.041]
53 Ahmad SS, Ahmad K, Lee EJ, Lee YH, Choi I. Implications of Insulin-Like Growth Factor-1 in Skeletal Muscle and Various Diseases.
2020; 9 [PMID: 32722232 DOI: 10.3390/cells9081773]
54 Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance.
2018; 98: 2133-2223[PMID: 30067154 DOI: 10.1152/physrev.00063.2017]
55 Bianchi VE, Locatelli V, Rizzi L. Neurotrophic and Neuroregenerative Effects of GH/IGF1.
2017; 18 [PMID:29149058 DOI: 10.3390/ijms18112441]
56 Pomytkin I, Costa-Nunes JP, Kasatkin V, Veniaminova E, Demchenko A, Lyundup A, Lesch KP, Ponomarev ED,Strekalova T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment.
2018; 24: 763-774 [PMID: 29691988 DOI: 10.1111/cns.12866]
57 Kleinridders A, Ferris HA, Cai W, Kahn CR. Insulin action in brain regulates systemic metabolism and brain function.
2014; 63: 2232-2243 [PMID: 24931034 DOI: 10.2337/db14-0568]
58 Hakuno F, Takahashi SI. IGF1 receptor signaling pathways.
2018; 61: T69-T86 [PMID: 29535161 DOI:10.1530/JME-17-0311]
59 Apostolatos A, Song S, Acosta S, Peart M, Watson JE, Bickford P, Cooper DR, Patel NA. Insulin promotes neuronal survival
the alternatively spliced protein kinase CδII isoform.
2012; 287: 9299-9310 [PMID: 22275369 DOI: 10.1074/jbc.M111.313080]
60 Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes?
2005; 7: 63-80 [PMID: 15750215 DOI: 10.3233/jad-2005-7107]
61 Morris JK, Vidoni ED, Perea RD, Rada R, Johnson DK, Lyons K, Pahwa R, Burns JM, Honea RA. Insulin resistance and gray matter volume in neurodegenerative disease.
2014; 270: 139-147 [PMID: 24735819 DOI:10.1016/j.neuroscience.2014.04.006]
62 Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology?
2010; 6: 551-559 [PMID: 20842183 DOI: 10.1038/nrneurol.2010.130]
63 Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling.
2010; 31: 224-243 [PMID: 18479783 DOI: 10.1016/j.neurobiolaging.2008.04.002]
64 Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey WH 2nd, Craft S. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults.
2008; 13: 323-331 [PMID: 18430999 DOI: 10.3233/jad-2008-13309]
65 Del Campo M, Stargardt A, Veerhuis R, Reits E, Teunissen CE. Accumulation of BRI2-BRICHOS ectodomain correlates with a decreased clearance of Aβ by insulin degrading enzyme (IDE) in Alzheimer's disease.
2015; 589: 47-51 [PMID: 25597881 DOI: 10.1016/j.neulet.2015.01.036]
66 Toral-Rios D, Pichardo-Rojas PS, Alonso-Vanegas M, Campos-Peña V. GSK3β and Tau Protein in Alzheimer's Disease and Epilepsy.
2020; 14: 19 [PMID: 32256316 DOI: 10.3389/fncel.2020.00019]
67 Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, Kiosea NC, Nazari S, Chen PP, Nothias F, Chan P, Teng E,Frautschy SA, Cole GM. Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging.
2014; 34: 7124-7136 [PMID: 24849348 DOI: 10.1523/JNEUROSCI.3439-13.2014]
68 Chatterjee S, Mudher A. Alzheimer's Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits.
2018; 12: 383 [PMID: 29950970 DOI: 10.3389/fnins.2018.00383]
69 Mullins RJ, Diehl TC, Chia CW, Kapogiannis D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer's Disease.
2017; 9: 118 [PMID: 28515688 DOI: 10.3389/fnagi.2017.00118]
70 Dienel GA. Brain Glucose Metabolism: Integration of Energetics with Function.
2019; 99: 949-1045 [PMID:30565508 DOI: 10.1152/physrev.00062.2017]
71 Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation.
2010; 219: 25-32 [PMID: 20004026 DOI: 10.1016/j.jneuroim.2009.11.010]
72 Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, Farhang-Fallah J, Dikkes P, Warot XM, Rio C, Corfas G,White MF. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation.
2003; 23: 7084-7092 [PMID: 12904469 DOI: 10.1523/JNEUROSCI.23-18-07084.2003]
73 Johnson LA, Torres ER, Impey S, Stevens JF, Raber J. Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome.
2017; 7: 43701 [PMID: 28272510 DOI: 10.1038/srep43701]
74 Athauda D, Foltynie T. Protective effects of the GLP-1 mimetic exendin-4 in Parkinson's disease.
2018; 136: 260-270 [PMID: 28927992 DOI: 10.1016/j.neuropharm.2017.09.023]
75 Marciniak E, Leboucher A, Caron E, Ahmed T, Tailleux A, Dumont J, Issad T, Gerhardt E, Pagesy P, Vileno M,Bournonville C, Hamdane M, Bantubungi K, Lancel S, Demeyer D, Eddarkaoui S, Vallez E, Vieau D, Humez S, Faivre E,Grenier-Boley B, Outeiro TF, Staels B, Amouyel P, Balschun D, Buee L, Blum D. Tau deletion promotes brain insulin resistance.
2017; 214: 2257-2269 [PMID: 28652303 DOI: 10.1084/jem.20161731]
76 Liu P, Cui L, Liu B, Liu W, Hayashi T, Mizuno K, Hattori S, Ushiki-Kaku Y, Onodera S, Ikejima T. Silibinin ameliorates STZ-induced impairment of memory and learning by up- regulating insulin signaling pathway and attenuating apoptosis.
2020; 213: 112689 [PMID: 31669775 DOI: 10.1016/j.physbeh.2019.112689]
77 Grizzanti J, Corrigan R, Servizi S, Casadesus G. Amylin Signaling in Diabetes and Alzheimer's Disease: Therapy or Pathology?
2019; 4: 12-16 [PMID: 31511851 DOI: 10.29245/2572.942X/2019/1.1212]
78 Zhu H, Tao Q, Ang TFA, Massaro J, Gan Q, Salim S, Zhu RY, Kolachalama VB, Zhang X, Devine S, Auerbach SH,DeCarli C, Au R, Qiu WQ. Association of Plasma Amylin Concentration With Alzheimer Disease and Brain Structure in Older Adults.
2019; 2: e199826 [PMID: 31433485 DOI: 10.1001/jamanetworkopen.2019.9826]
79 Ly H, Verma N, Sharma S, Kotiya D, Despa S, Abner EL, Nelson PT, Jicha GA, Wilcock DM, Goldstein LB, Guerreiro R,Brás J, Hanson AJ, Craft S, Murray AJ, Biessels GJ, Troakes C, Zetterberg H, Hardy J, Lashley T, Aesg, Despa F. The association of circulating amylin with β-amyloid in familial Alzheimer's disease.
2021; 7: e12130[PMID: 33521236 DOI: 10.1002/trc2.12130]
80 Martinez-Valbuena I, Valenti-Azcarate R, Amat-Villegas I, Riverol M, Marcilla I, de Andrea CE, Sánchez-Arias JA, Del Mar Carmona-Abellan M, Marti G, Erro ME, Martínez-Vila E, Tuñon MT, Luquin MR. Amylin as a potential link between type 2 diabetes and alzheimer disease.
2019; 86: 539-551 [PMID: 31376172 DOI: 10.1002/ana.25570]
81 Petrus AT, Lighezan DL, Danila MD, Duicu OM, Sturza A, Muntean DM, Ionita I. Assessment of platelet respiration as emerging biomarker of disease.
2019; 68: 347-363 [PMID: 30904011 DOI: 10.33549/physiolres.934032]
82 Ortiz GG, Pacheco Moisés FP, Mireles-Ramírez M, Flores-Alvarado LJ, González-Usigli H, Sánchez-González VJ,Sánchez-López AL, Sánchez-Romero L, Díaz-Barba EI, Santoscoy-Gutiérrez JF, Rivero-Moragrega P. Oxidative Stress:Love and Hate History in Central Nervous System.
2017; 108: 1-31 [PMID: 28427557 DOI:10.1016/bs.apcsb.2017.01.003]
83 Serrano F, Klann E. Reactive oxygen species and synaptic plasticity in the aging hippocampus.
2004; 3:431-443 [PMID: 15541710 DOI: 10.1016/j.arr.2004.05.002]
84 Huang TJ, Price SA, Chilton L, Calcutt NA, Tomlinson DR, Verkhratsky A, Fernyhough P. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia.
2003; 52: 2129-2136 [PMID: 12882932 DOI: 10.2337/diabetes.52.8.2129]
85 Moreira PI, Santos MS, Sena C, Seiça R, Oliveira CR. Insulin protects against amyloid beta-peptide toxicity in brain mitochondria of diabetic rats.
2005; 18: 628-637 [PMID: 15755688 DOI: 10.1016/j.nbd.2004.10.017]
86 Santos RX, Correia SC, Alves MG, Oliveira PF, Cardoso S, Carvalho C, Duarte AI, Santos MS, Moreira PI. Insulin therapy modulates mitochondrial dynamics and biogenesis, autophagy and tau protein phosphorylation in the brain of type 1 diabetic rats.
2014; 1842: 1154-1166 [PMID: 24747740 DOI: 10.1016/j.bbadis.2014.04.011]
87 Ruegsegger GN, Vanderboom PM, Dasari S, Klaus KA, Kabiraj P, McCarthy CB, Lucchinetti CF, Nair KS. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain.
2019; 4 [PMID: 31534057 DOI: 10.1172/jci.insight.130681]
88 Carvalho C, Santos MS, Oliveira CR, Moreira PI. Alzheimer's disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers.
2015; 1852: 1665-1675 [PMID: 25960150 DOI:10.1016/j.bbadis.2015.05.001]
89 Kleinridders A, Cai W, Cappellucci L, Ghazarian A, Collins WR, Vienberg SG, Pothos EN, Kahn CR. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders.
2015; 112: 3463-3468 [PMID:25733901 DOI: 10.1073/pnas.1500877112]
杂志排行

World Journal of Diabetes的其它文章
- Risk factors for mortality within 6 mo in patients with diabetes undergoing urgent-start peritoneal dialysis: A multicenter retrospective cohort study
- Roles of transient receptor potential channel 6 in glucose-induced cardiomyocyte injury
- Long noncoding RNA X-inactive specific transcript regulates NLR family pyrin domain containing 3/caspase-1-mediated pyroptosis in diabetic nephropathy
- Gut microbiota and diabetic kidney diseases: Pathogenesis and therapeutic perspectives
- lnsulin-resistance in paediatric age: lts magnitude and implications