小鹅瘟病毒荧光RPA恒温快速检测方法的建立与应用
2022-05-16徐龙涛冯宗玲曲光刚李本科
高 远,徐龙涛,冯宗玲,曲光刚,李本科
(1.荣成市畜牧业发展中心,山东荣成 264399;2.齐鲁动物保健品有限公司,济南 250100;3.滨州市科技创新发展研究院,山东滨州 256600;4.山东省滨州畜牧兽医研究院,山东滨州 256600;5.滨州市农业技术推广中心,山东滨州 256600)
小鹅瘟是一种常见的水禽急性或亚急性败血性传染病,临床特征主要表现为厌食、渴欲增强、无力、不愿活动,常表现出摇头症状[1]。该病的发病病程短,传染性强,死亡率高,可高达90%~100%,但发病率和致死率随着雏鹅的日益长大而下降[2]。该病由小鹅瘟病毒(Goose parvovirus,GPV)引起。1956年我国学者方定一在扬州首次发现并分离到第一株小鹅瘟病毒,1961年他又用鹅胚分离到一株与鸡新城疫及鸭肝炎病毒无关的新病毒,并将该病命名为小鹅瘟,病原体称为小鹅瘟病毒[3]。此后许多国家都报道过该疾病,包括英国和瑞典等,对全球的养鹅业造成了严重危害和巨大的经济损失。GPV主要感染雏鹅和番鸭,其它家禽能够天然抵抗该病毒的感染[4]。GPV属于细小病毒科、细小病毒属,是一种无包膜的单链DNA病毒,基因组长度约为5100 bp,包含两个开放阅读框(ORF):位于5’端的ORF1编码参与病毒复制的非结构蛋白和调控蛋白,位于3’端的ORF2编码结构蛋白VP1、VP2和VP3。表面结构蛋白表现出病毒的遗传变异,编码这些结构蛋白的基因突变改变了病毒的免疫特性。由于VP2和VP3基因区域同时包含保守区和可变区而被专门用于检测病毒进化和遗传变异。
目前,GPV的检测方法多种多样,主要包括病原学检测、血清学检测和分子生物学检测。病原学检测方法主要是病毒的分离鉴定[5],是检测小鹅瘟最经典和最准确的方法之一,但是该方法检测所需时间长,操作复杂,不满足小鹅瘟临床快速检测的需求。血清学检测主要包括琼脂免疫扩散试验[6]、病毒中和试验[7]、酶联免疫吸附试验[8]、反向间接血凝试验[9]、免疫荧光抗体法[10]、免疫过氧化物酶染技术[11]、对流免疫电泳法[12]和精子凝集抑制试验[13]等,但是上述方法均存在不同的缺点,或检测灵敏度低,或检测时间长,或操作复杂等。分子生物学检测主要有PCR检测、核酸探针技术、荧光定量PCR检测和环介导等温扩增(LAMP)等。2006年,李福伟等[14]建立了检测GPV的PCR检测方法,并针对保守序列VP3设计了一对特异性引物,成功扩增出GPV VP3基因的目的片段,敏感性可达到8 pg。2005年,布日额等[15]建立了另一种核酸探针方法,以SCPD为底物的化学发光的地高辛标记核酸探针检测GPV,该方法特异性、敏感性和适用性都很强,但其技术性要求较强,操作繁琐,且检测成本较高,不适宜临床上的推广与应用。2010年,龙朕等[16]根据GPV的保守序列NS基因建立了荧光PCR检测方法,结果表明,该方法具有良好的敏感性和特异性,但是该方法需要荧光定量PCR仪,不方便携带,不满足现场检测的需求。2012年,金文杰等[17]建立了LAMP技术应用于GPV的快速诊断,缩短了检测时间,但是该方法不仅需要设计三对特异性引物,而且极易出现假阳性。
重组酶聚合酶扩增技术(Recombinase polymerase amplification, RPA)是2006年由英国科学家研发的一种核酸恒温扩增技术。它是利用多种酶参与,如重组酶、单链结合蛋白、外链核酸酶III和链置换DNA聚合酶,在体外模拟生物体内DNA复制,恒温条件下短时间内实现靶基因大量扩增。重组酶与引物结合形成复合物,在双链DNA中寻找同源序列,一旦定位到同源序列,就会发生链交换反应,形成并启动DNA合成,模板上的靶区进行指数级扩增,被取代的DNA链与单链结合蛋白结合,防止进一步的替代。RPA技术自2006年首次引入我国便受到了广泛关注,随后出现了大量关于该技术的研究,近年来取得显著进展[18-19]。RPA技术可在37~42 ℃恒温条件下快速扩增,DNA扩增到可检测水平所需的时间取决于样品的DNA拷贝数,结果一般可以在20 min内观察到。RPA检测对样品制备要求低,具有恒温、快速、便携、灵敏度高、特异性强和操作简单等优点[20],是目前有望替代PCR技术的核酸等温扩增方法,具有广阔的应用前景。本研究以VP3基因为目的基因建立了检测GPV的RPA恒温快速检测方法。
1 材料与方法
1.1 病毒核酸及临床样本 鹅副黏病毒、鹅源鸭瘟病毒、小鹅流行性感冒病毒、鹅副伤寒病毒、大肠杆菌、曲霉菌和GPV均保存于齐鲁动保实验室;小鹅瘟临床病料由山东养鹅厂提供。
1.2 GPV标准质粒的构建 根据GenBank公布的GPV的全基因组序列,利用分子生物学软件primer premier 5.0,针对VP3保守区域设计一对特异性引物(表1)。引物由上海生物工程技术服务有限公司合成。

表1 GPV扩增引物
根据设计的引物扩增出1605 bp的VP3片段,并构建重组质粒GPV-VP3-18T,转化至DH5α大肠杆菌感受态细胞,经菌液PCR鉴定阳性菌株,使用质粒提取Mini试剂盒Ⅰ(OMEGA,USA)提取质粒DNA。用NanoDrop-2000分光光度计(NanoDrop, Wilmington, USA)定量质粒DNA的浓度,将重组质粒送上海生工测序鉴定。
1.3 荧光RPA恒温快速检测方法引物与探针的设计 根据GenBank收录的GPV的VP3序列,利用primer premier 5.0软件,根据RPA引物设计原则,设计GPV VP3基因特异性的引物和探针(表2),并由上海生物工程技术服务有限公司合成。

表2 GPV的RPA引物和探针
1.4 核酸的提取及反转录 使用AXYGEN试剂盒(AXYGEN,美国)提取GPV、鹅副黏病毒、鹅源鸭瘟病毒、小鹅流行性感冒病毒、鹅副伤寒病毒的核酸,用20 μL无核酸酶水洗脱病毒中的核酸,使用NanoDrop-2000分光光度计测定病毒核酸的浓度。使用细菌基因组 DNA提取试剂盒(康为世纪,中国)提取大肠杆菌和曲霉菌的基因组DNA。所有核酸样品提取完成后,-20 ℃保存备用。
采用反转录试剂盒TransScript®Ⅱ One-Step gDNA Removal and cDNA Synthesis SuperMix去除基因组DNA并合成鹅副黏病毒与小鹅流行性感冒病毒cDNA。在Rase Free Microtube管中加入7.0 μL模板RNA、1.0 μL下游引物、1.0 μL TransScript®Ⅱ RT/RI Enzyme Mix、1.0 μL gDNA Remover、10.0 μL 2×TS Ⅱ Reaction Mix配制溶液。混合液在50 ℃孵育30 min后,85 ℃加热5 s。得到的cDNA溶液于-20 ℃保存备用。
1.5 荧光RPA恒温扩增方法的建立 荧光RPA恒温扩增方法反应体系为:溶解剂10 μL,上游引物1 μL,下游引物1 μL,探针0.3 μL,激活剂1 μL,模板1 μL,加入去离子水至25 μL;最佳反应条件为:40 ℃条件下反应20 min。除激活剂和模板外,其余以混合溶液的形式加入含有冻干粉的0.2 mL EP管中,涡旋混合离心。将核酸加到反应管中,将激活剂加到反应管盖上。上下颠倒混合后,瞬时离心;离心后,荧光值由恒温荧光检测仪读取,每30 s监测一次荧光值。以重组质粒GPV-VP3-18T为模板,以RNase游离水为阴性对照,对该方法的引物浓度、反应温度和反应时间等条件进行优化,具体操作如下:利用同一探针,上下游引物采用3个浓度梯度(0.6 μmol/L、0.8 μmol/L、1 μmol/L),确定最佳引物浓度;利用最佳引物浓度采用3个反应梯度温度(38 ℃、40 ℃、42 ℃),确定该方法的最佳反应温度。荧光值由恒温荧光检测仪读取,每30 s监测一次荧光值。恒温荧光检测仪检测结果为阳性且出现标准S型曲线判为检测出阳性结果。
1.6 RPA的敏感性和特异性测定 为测定建立的荧光RPA检测方法的敏感性,将已构建的重组质粒DNA进行连续的10倍稀释,作为荧光RPA检测方法敏感性试验的标准品。每个稀释浓度的标准品加入1 μL作为反应模板进行RPA反应扩增以确定该方法的最低检测拷贝数。同时,以鹅常见疾病不同病原体基因组DNA或RNA作为模板分析该荧光RPA检测方法的特异性。采用优化后的方法分别对鹅副黏病毒cDNA、鹅源鸭瘟病毒DNA、小鹅流行性感冒病毒cDNA、鹅副伤寒病毒DNA、大肠杆菌DNA和曲霉菌DNA进行检测,以GPV的阳性样品为阳性对照,以无菌水作为阴性对照,根据检测结果确定该方法的特异性。
1.7 RPA的重复性测定 分别以浓度为1×103、1×102、1×101copies/μL的GPV-VP3-18T标准质粒为模板进行RPA扩增,每个稀释度重复三次,根据其Ct值计算变异系数,评价该方法的重复性。
1.8 RPA、PCR和Real-time PCR的敏感性比较 对已建立的GPV PCR[21]和GPV Real-time PCR[22]检测方法与本研究建立的GPV RPA检测方法进行敏感性比较。使用AXYGEN试剂盒提取GPV的基因组DNA并将提取的核酸进行10倍倍比稀释作为模板,同时用PCR、Real-time PCR和RPA三种检测方法进行检测,比较三种方法的检测敏感度。
1.9 临床病料的检测 从山东省不同的养殖场或农户采集了100份疑似组织、血液和体液样本,使用AXYGEN试剂盒提取核酸,采用本研究建立的RPA方法和Real-time PCR方法进行检测,分析两种方法的符合率。
2 结果与分析
2.1 GPV VP3基因PCR扩增 通过PCR方法扩增靶标基因VP3,PCR产物进行凝胶电泳成像(图1),结果表明本研究成功扩增出GPV VP3目的条带,目的片段大小约为1605 bp,与预期大小一致。
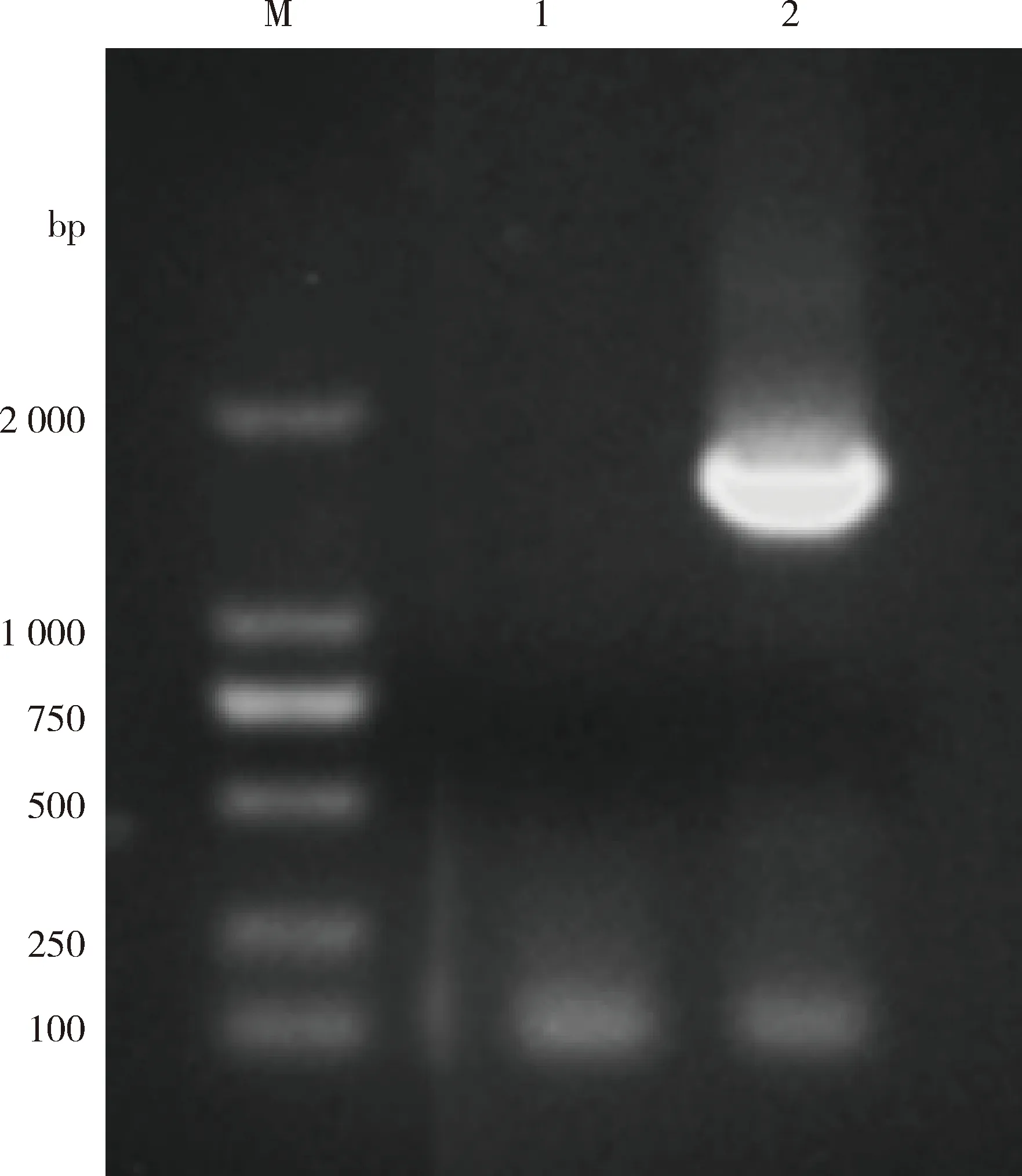
M:DL-2000 DNA分子质量标准;1:阴性对照;2:PCR扩增产物
2.2 菌液PCR鉴定结果 挑取单菌落过夜培养后进行菌液PCR鉴定,泳道6的阴性结果成立,泳道1-5的结果为GPV阳性(图2),根据结果将检测为阳性菌液的样本进行质粒提取并测序。测序结果显示5个阳性质粒插入片段均为目的基因,表明VP3基因标准质粒构建成功。

M:DL-2000 DNA分子质量标准;1-5:菌液样品;6:阴性对照
2.3 引物的筛选 所有设计的GPV引物组合均有S型的扩增曲线,其中GPV-F1和GPV-R1组合所得的荧光值为4000,Ct值为7.0(图3),均为最佳,故确定最佳引物组合为GPV-F1和GPV-R1。
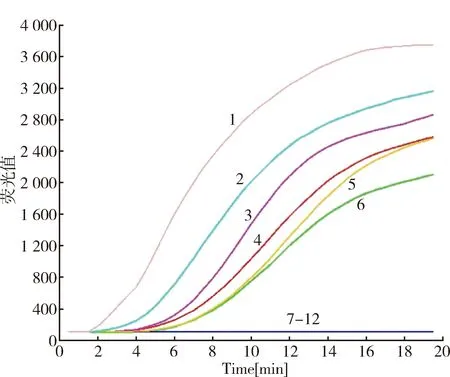
1-6:引物GPV-F1-R1、GPV-F3-R1、GPV-F2-R1、GPV-F1-R2、GPV-F2-R2、GPV-F3-R2;7-12:引物阴性对照GPV-F1-R1、GPV-F1-R2、GPV-F2-R1、GPV-F2-R2、GPV-F3-R1、GPV-F3-R2 1-6:Primers GPV-F1-R1、GPV-F3-R1、GPV-F2-R1、GPV-F1-R2、GPV-F2-R2、GPV-F3-R2;7-12:Negative control of primers GPV-F1-R1、GPV-F1-R2、GPV-F2-R1、GPV-F2-R2、GPV-F3-R1、GPV-F3-R2
2.4 引物浓度优化 测试引物浓度分别为0.6 μmol/L、0.8 μmol/L、1 μmol/L,其中引物浓度为1 μmol/L时出峰时间最早,且其荧光值最高(图4),故最佳引物浓度选择1 μmol/L。
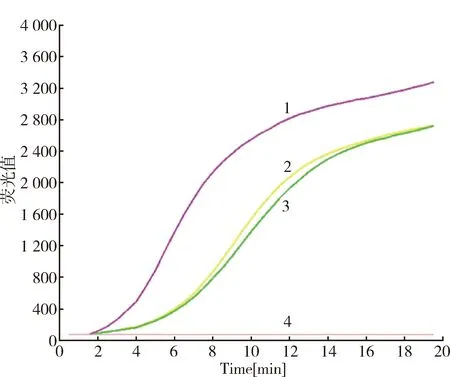
1: 1 μmol/L primers; 2: 0.8 μmol/L primers; 3: 0.6 μmol/L primers; 4: Negative control
2.5 反应温度优化 GPV RPA方法采用的反应温度分别为38 ℃、40 ℃、42 ℃,其中40 ℃出峰时间最早,Ct值为6.0,其荧光值最高,为4500(图5),故最佳反应温度确定为40 ℃。

A: 38 ℃; B: 40 ℃; C: 42 ℃
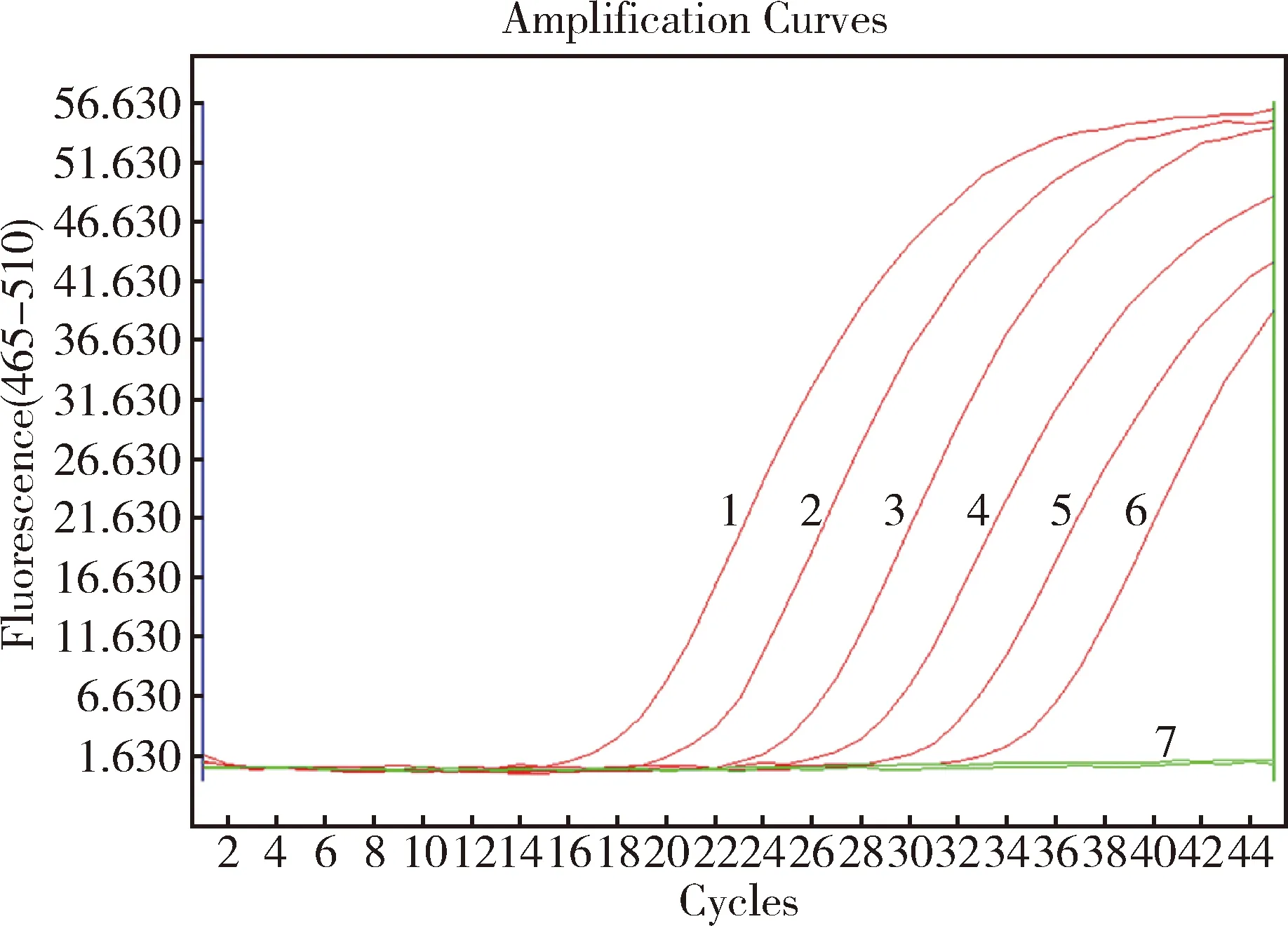
2.6 敏感性和特异性试验结果 GPV RPA检测方法只扩增出GPV核酸,其他病毒或细菌的核酸均为阴性,无交叉反应,说明该方法具有很好的特异性(图6)。GPV荧光RPA检测方法最低能够检测到10 copies/μL的标准质粒(图7)。

1:GPV病毒;2:阴性对照3:鹅副黏病毒;4:鹅源鸭瘟病毒;5:小鹅流行性感冒病毒;6:鹅副伤寒病毒;7:大肠杆菌;8:曲霉菌

1: 105copies/μL; 2: 104copies/μL; 3: 103copies/μL; 4: 102copies/μL; 5: 101copies/μL; 6: 100copies/μL; 7: Negative control
2.7 重复性实验结果 以三种不同拷贝数的标准质粒为模板进行三次重复实验来验证该方法的重复性,拷贝数为1000 copies/μL时,Ct值为7.3、7.3、8.0,变异系数为5.36%,荧光值为4600、4500、4500,重复性良好(图8A);拷贝数为100 copies/μL时,Ct值为9.3、9.3、9.0,变异系数为1.88%,荧光值为4000、3900、3850,重复性良好(图8B);拷贝数为10 copies/μL时Ct值为10.0、10.0、10.3,变异系数为1.71%,荧光值为2600、2700、2500,重复性良好(图8C)。结果表明该方法重复性良好。

A:模板拷贝数为1000 copies/μL;B:模板拷贝数为100 copies/μL;C:模板拷贝数为10 copies/μL
2.8 RPA、PCR和Real-time PCR的敏感性比对实验结果 按照已建立的检测条件,用相同稀释倍数的核酸作为模板同时利用PCR、Real-time PCR和RPA三种方法进行检测。其中,PCR检测方法能够检测到的核酸稀释倍数为103(图9), Real-time PCR检测方法能够检测到的核酸稀释倍数为105(图10),而RPA检测方法能够检测到的核酸稀释倍数为104(图11)。

1:100稀释;2:101稀释;3:102稀释;4:103稀释;5:104稀释;6:105稀释;M:DL-2000 DNA Marker
1:100稀释;2:101稀释;3:102稀释;4:103稀释;5:104稀释;6:105稀释;7:阴性对照
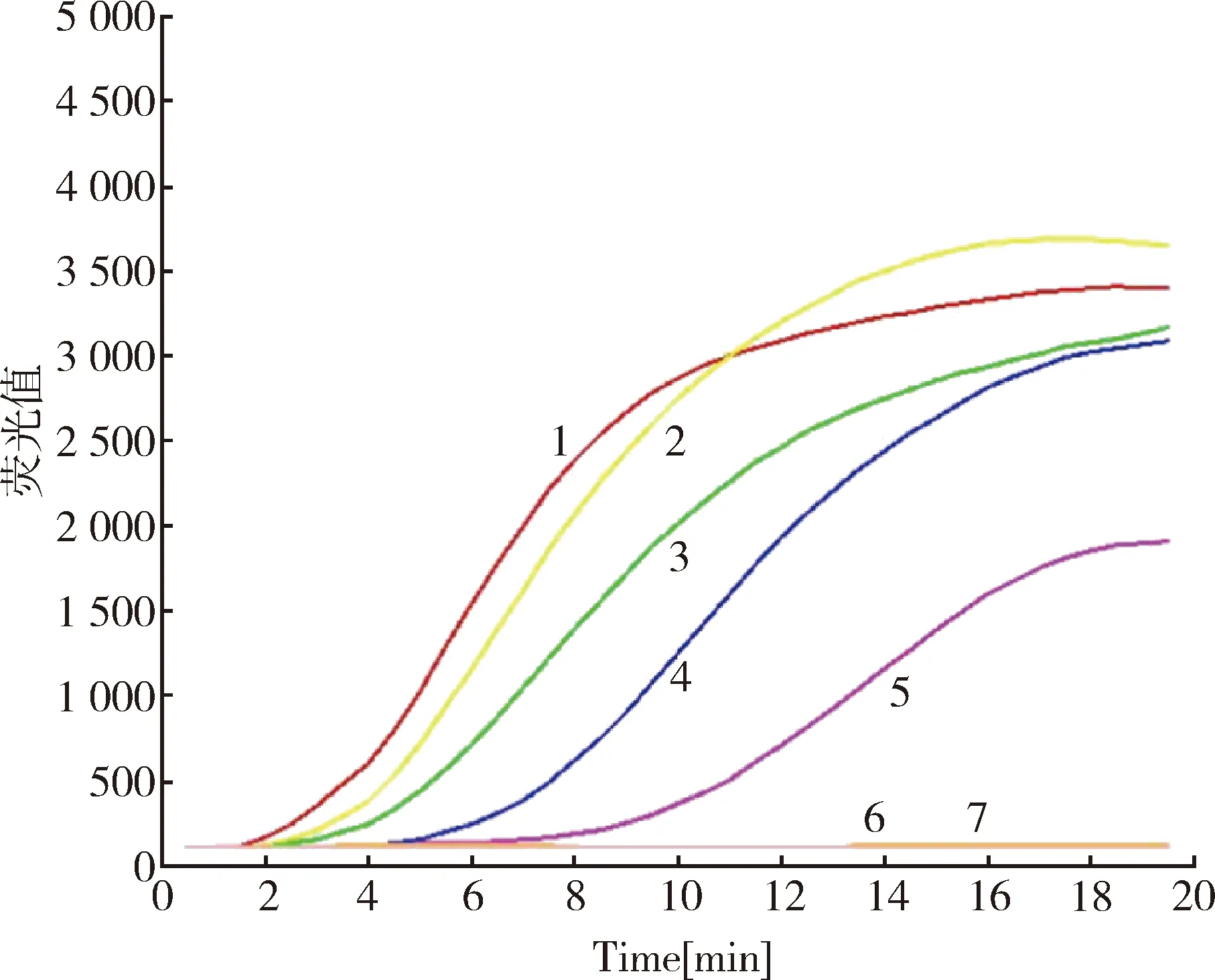
1:100稀释;2:101稀释;3:102稀释;4:103稀释;5:104稀释;6:105稀释;7:阴性对照
2.9 临床病料的检测结果 对收集到的100个临床疑似样本同时采用已建立的RPA方法和Real-time PCR进行检测,结果见表3,两种方法的符合率为99%。
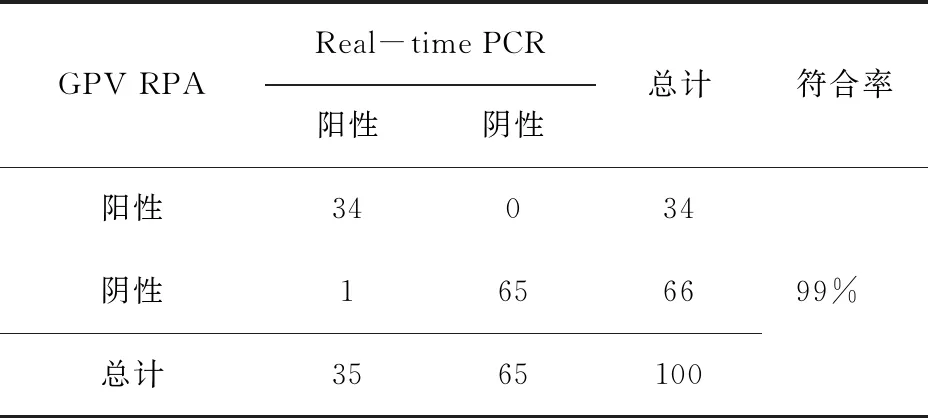
表3 GPV RPA与Real-time PCR方法临床样本检测符合性试验结果
3 讨 论
GPV主要感染各种鹅、番鸭和莫斯科鸭,该病毒对幼鹅致死率可达100%,对大日龄鹅呈亚临床感染从而传染给幼鹅,给养鹅行业造成巨大损失。因此,GPV的快速诊断是预防和控制该病的重要前提。本研究采用了一种基于EXO探针的RPA检测方法来检测GPV,可以在40 ℃恒温条件下20 min内完成检测,检测限可达10 copies/μL,仅可检测GPV,变异系数低于6%,具有良好的特异性和重复性;与Real-time PCR方法的符合率达到99%。
根据已报道的GPV PCR[21]和Real-time PCR[22]检测方法,比较了RPA与该两种方法的敏感性。结果表明,RPA的敏感性比PCR高10倍,但比Real-time PCR低10倍(图9-图11)。与环介导等温扩增(LAMP)[17]检测方法检测GPV相比,LAMP检测需要3对引物、更高的温度(62 ℃)和更长的运行时间,而RPA检测方法仅需要一对引物和一个探针,因此,该方法更简单,所需时间更短。
本研究建立的GPV RPA方法有以下优点:首先,RPA试剂以冻干粉的形式制备,室温下可保存12周而不失去活性,与其他试剂相比,RPA试剂稳定。其次,RPA快速检测方法在恒温条件下进行,不需要昂贵的热循环器,成本更低。第三,合成的RPA引物和探针可容许有5~9个错配,对实验性能没有影响,而Real-time PCR错配会导致探针失效或失去灵敏度。第四,PRA反应对常见的PCR抑制剂有一定的耐受性,从而保证了反应的稳定性。
本研究成功建立了一种GPV的RPA恒温检测方法,对GPV现场检测试剂盒的开发具有重要意义。
