IgG4相关腹膜后纤维化1例报告
2022-05-14李朝霞纪竹慧辛桂杰
李朝霞, 刘 洋, 李 楠, 纪竹慧, 辛桂杰
吉林大学第一医院 肝胆胰内科, 长春 130021
IgG4相关性疾病(IgG4-related disease,IgG4-RD)是一种由免疫机制失衡介导,累及多器官或组织并伴随组织淋巴细胞浸润及纤维化的慢性进行性自身免疫病,发病率低,可累及胰腺、腹膜后、唾液腺、颌下腺、胆管等多种器官[1],临床极易误诊,部分患者可合并腹膜后纤维化(retroperitoneal fibrosis,RPF),则更易误诊,大约10%的IgG4-RD患者可合并腹膜后纤维化[2],即IgG4相关RPF IgG4-RPF。该病罕见,国内外对其报道较少,现报道IgG4-RPF且糖皮质激素治疗成功1例。
1 病例资料
患者男性,56岁,主因“腹痛1个月”于2020年4月4日入本院。1个月前无明显诱因出现上腹部疼痛,为间断性钝痛,与进食无关。当地胃镜检查示“非萎缩性胃炎伴糜烂、十二指肠球炎、贲门口炎”,口服泮托拉唑2周无好转,腹部CT示胰腺占位性病变,遂入本院。既往体健,无特殊病史。入院查体:皮肤、巩膜无黄染,未见肝掌及蜘蛛痣,双侧颈部及颌下淋巴结触及肿大,界清,质硬,无压痛,活动度好,大小约1.2 cm。腹软,中上腹压痛,无反跳痛及肌紧张,未触及腹部包块,肝、脾肋下未触及。辅助检查:胰酶LPS 168 U/L;肝功能:AST 86.6 U/L,ALT 111.0 U/L,GGT 440.6 U/L,ALP 230.0 U/L,Alb 35.8 g/L;CRP 7.20 mg/L;肿瘤标志物、血常规、凝血常规、尿常规、便常规、血清HBsAg、抗-HBs、HBeAg、抗-HBe、抗-HBc、IgM、IgA、C3、C4、ANA系列、ANCA确证-PR3,MPO,GBM+ANCA筛查均未见异常;IgG4 6.020 g/L,IgE 2020.00 IU/ml。全腹CT平扫+三期增强(图1)示:(1)肝内胆管略扩张,肝门区胆管走行区软组织影,考虑占位性病变;(2)胰尾部占位性病变(约4.1×3.0 cm),不除外恶性;(3)腹腔干周围软组织影(约3.1 cm×3.5 cm),左中腹肠系膜区多发结节影、团块影,待除外转移;腹主动脉CTA(图2)示:(1)腹腔干、肠系膜上动脉周围低密度影;(2)左上腹多发软组织密度影,包绕肠系膜上动脉部分远端分支;(3)肝右叶部分胆管扩张,胰尾占位病变。PET-CT示:(1)胰腺尾部高代谢灶、肝门区胆管走形高代谢灶、多处高代谢淋巴结(左腹部肠系膜区)、腹腔干周围高代谢灶、腹主动脉及右侧髂血管管壁局限壁厚伴代谢增高,均考虑IgG4相关疾病;(2)双侧颈部、颌下淋巴结炎性增生;(3)肝内胆管及胆总管轻度扩张。本例患者以腹痛为主症,查体双侧颈部及颌下淋巴结触及肿大,中上腹压痛,腹部影像学提示胰腺占位性病变,腹腔干周围存在软组织影,双侧颈部、颌下淋巴结炎性增生,考虑IgG4相关疾病可能性大,IgE 2020.00 IU/ml,IgG4为6.020 g/L,患者未同意行组织学检查,与患者及家属充分沟通病情后,予以初始口服醋酸泼尼松片30 mg/d,同时给予保肝、抑酸及对症支持治疗,治疗1周后腹痛较前明显减轻,双侧颈部及颌下淋巴结明显缩小,大小约0.8 cm,LPS 140 U/L,肝功能:AST 25.2 U/L,ALT 50.4 U/L,GGT 236.3 U/L,ALP 193.9 U/L。全腹CT平扫:腹腔干动脉周围软组织影较前变小(约2.3 cm×2.5 cm),左上腹多发结节影均较前略变小,肝总管、胰尾部改变较前相仿。患者激素治疗敏感有效,明确诊断:IgG4相关自身免疫性胰腺炎、IgG4-RPF。继续口服醋酸泼尼松片30 mg/d,激素治疗3周后复查腹部CT示腹腔干动脉周围软组织影(约2.3 cm×1.6 cm),左上腹多发结节影,均较前片变小,激素减量为25 mg/d(每两周递减5 mg),减量至15 mg后维持4周,激素治疗12周后复查腹部CT示腹腔干动脉周围软组织影(最宽处约1.4 cm),左上腹多发结节影较前片均变小。再次减量为10 mg/d维持4个月,激素治疗16周后复查腹部CT未见明显异常(腹腔干动脉周围软组织影及左上腹多发结节影均消失,胰腺形态恢复正常)。
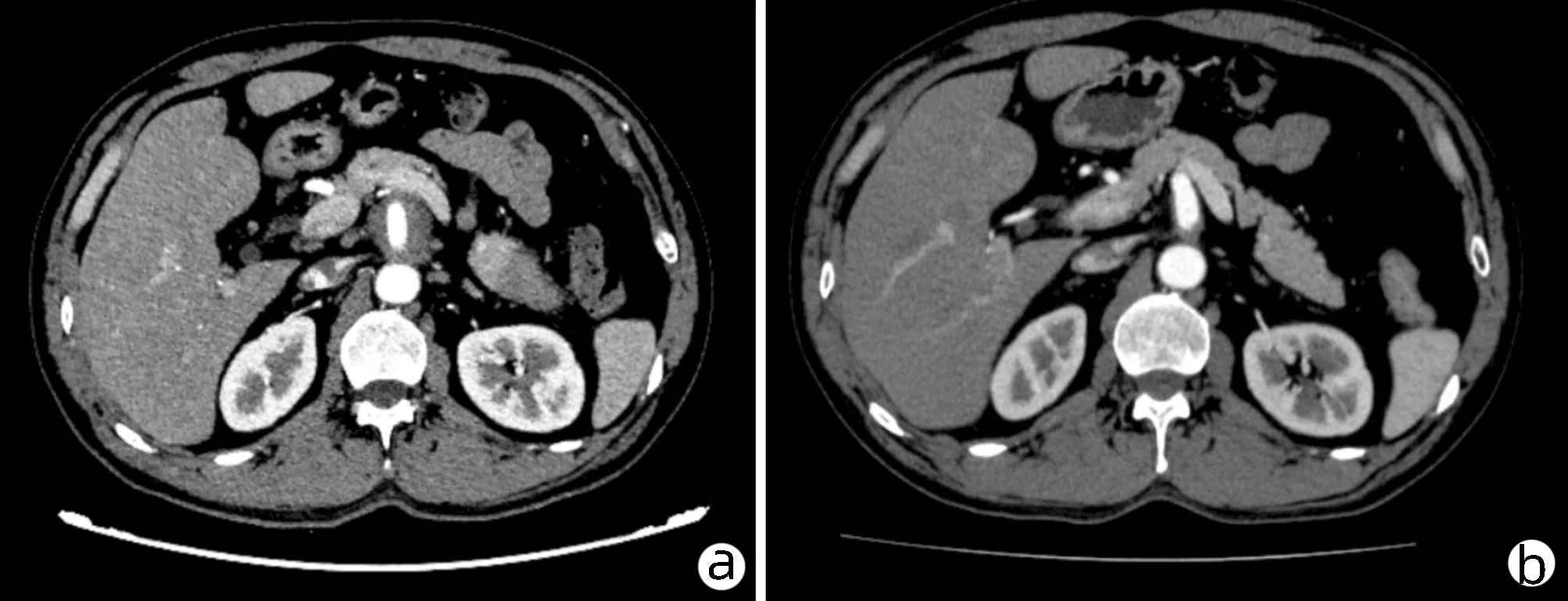
注:a,腹腔干周围见软组织密度影包绕,大小约3.1 cm×3.5 cm;b,腹腔干壁略厚,腺形态密度未见异常。
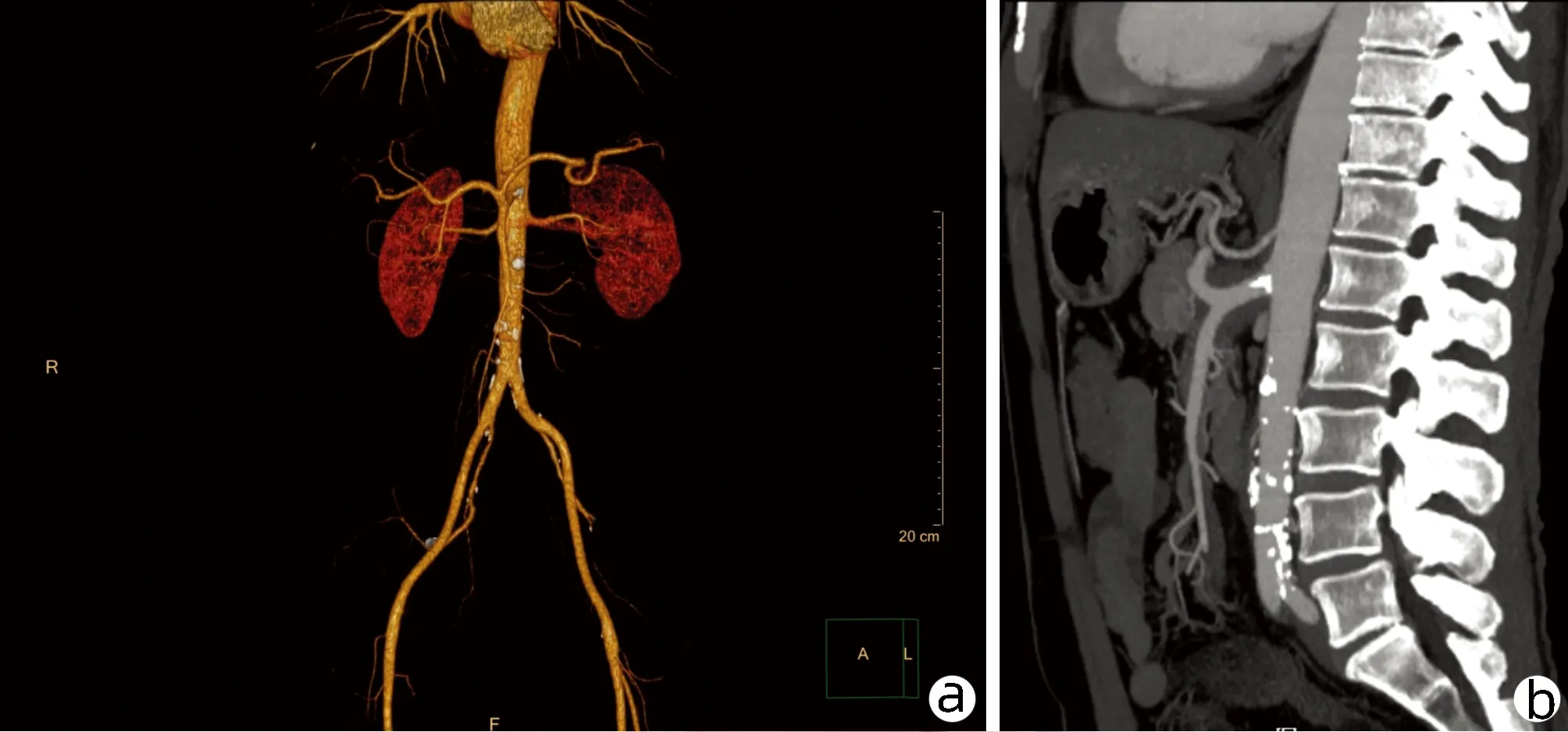
注:a,血管未见明显异常;b,腹腔干、肠系膜上动脉周围可见斑片状低密度影,累及长度约4.5 cm。
2 讨论
IgG4-RD是近年新发现的疾病,由Yoshida于1995年首次提出,随后Hamano、Kamisawa等相继报道类似病例[3-5],可累及胰腺、腹膜后、肾脏、胆管、唾液腺等多种组织及器官,其统一命名及诊断标准由日本IgG4-RD研究小组于2011年明确提出[6],日本报道其发病率为(0.28~1.08)/100 000,我国尚缺乏相应数据。发病机制目前认为主要与免疫机制失衡介导相关,可累及多种组织及器官,最常累及部位为胰腺,主要表现为胰腺弥漫性或局灶性肿胀,常需与胰腺癌鉴别。AIP患者主胰管狭窄与扩张并存,胰腺癌多为主胰管、胆总管的突然截断,磁共振胰胆管造影可呈“双管征”,胰头癌强化方式为乏血供,易侵及周围血管及器官,激素治疗有助于鉴别诊断,活检为金标准。IgG4-RD也可发生腹膜后纤维化,即IgG4-RPF,临床更易误诊,其属于特发性腹膜后纤维化。RPF分为特发性腹膜后纤维化(idiopathic retroperitoneal fibrosis,iRPF)和继发性腹膜后纤维化(secondary retroperitoneal fibrosis,sRPF),继发性多见于恶性肿瘤、感染、药物、放射治疗和罕见的组织细胞疾病,如Erdheim-Chester病,约占RPF的30%[7]。而iRPF的发病机制尚不明确,多与其他自身免疫性疾病相关联[8],目前国内外尚缺乏iRPF的大型流行病学资料,有研究[9]显示IgG4-RPF约占iRPF患者的30%~60%。
IgG4-RPF起病隐匿,缺乏特异性临床表现,主要症状有腹痛、腰痛、乏力、食欲降低、体质量减轻等,常见的异常指标有ESR加快、CRP升高、白细胞升高等,可有IgG4、IgE的升高,部分患者也可合并肾功能不全,一项研究[10]提示IgG2可作为IgG4-RD的一个补充的血清标志物,其准确性与IgG4相当。目前诊断主要依靠影像学及组织学表现。CT表现为腹膜后软组织影包绕腹主动脉、髂动脉或输尿管等,MRI表现为病灶在T1WI呈低信号,T2WI在疾病早期呈高信号,晚期呈低信号,有相关文献提示18F-FDG PET可用于评价iRPF的活动性以及治疗后残余病灶的代谢性[11]。组织活检是诊断iRPF的金标准,典型组织学表现为纤维化和淋巴细胞、浆细胞、巨噬细胞组成的单个核细胞炎性浸润。免疫组织化学提示IgG4阳性浆细胞与IgG阳性浆细胞的比例>40%,且IgG4阳性浆细胞>10个/高倍视野。淋巴细胞、浆细胞大量浸润、席纹状纤维化及闭塞性静脉炎是IgG4-RD的主要病理特征。但因其取材困难及风险大,一般在药物治疗不佳或怀疑恶变的患者中进行。该病例病初误诊为胰腺癌,完善相关检查后考虑IgG4相关自身免疫性胰腺炎,经激素治疗后,患者腹痛明显减轻,颈部及颌下淋巴结、腹腔干及左上腹多发结节影均明显缩小至消失,胰腺形态恢复正常,明确临床诊断:IgG4相关自身免疫性胰腺炎、IgG4-RPF。对于IgG4-RD合并腹膜后纤维化者,临床极易误诊,需综合判断,避免延误治疗。
IgG4-RPF的一线治疗方案为糖皮质激素,通常起始剂量为0.5~ 1 mg·kg-1·d-1,缓 解 后 逐 渐 减 量 至 维 持 剂 量 2.5~10 mg/d。激素抵抗或无法耐受其相关副作用以及激素治疗后复发的患者,可换用或加用其他药物联合治疗,如环磷酰胺、甲氨蝶呤、吗替麦考酚酯、环孢素等免疫抑制剂,但其是否可加强激素的治疗效果目前仍不清楚。近年来相关文献显示,雌激素受体拮抗剂他莫昔芬因其具有抗炎、抗纤维化及抗血管生成活性,且较激素可能更易耐受,也可用于IgG4-RPF的治疗,但研究[12-13]显示,其疗效不及糖皮质激素,目前尚缺乏大样本的观察与统计。此外,有文献[14]报道利妥昔单抗(rituximab,RTX)可用于IgG4-RPF患者的治疗,RTX是针对存在于B淋巴细胞表面的CD20抗原的嵌合抗体,循环B淋巴细胞是产生 IgG4的浆细胞的前体,RTX可导致循环B淋巴细胞的消耗。对于梗阻及其他器官受压迫症状明显、激素起效缓慢的患者,可行外科手术,目前主要的手术方式包括双J管置入术、肾脏穿刺造瘘术等。本例患者经长达8个月的激素治疗后,腹部症状缓解,腹膜后软组织影消失。
IgG4-RD较为少见,而IgG4-RPF更为罕见,临床容易漏诊、误诊。因此,当临床遇到腹痛、腹膜后组织肿物,CT检查发现有异常密度灶包绕腹膜后大血管,胰腺形态改变,应想到该病的可能,并结合患者症状体征及相关检查结果,综合分析病情,仔细鉴别诊断。对于IgG4-RPF的患者,在无激素应用禁忌的前提下,应及时把握适应证尽早行激素治疗,但该病在激素减量或停药后较易复发,需长期随访并维持治疗。
伦理学声明:本例报告已获得患者知情同意。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:李朝霞负责课题设计,资料分析,撰写论文;刘洋、李楠、纪竹慧参与收集数据,修改论文;辛桂杰负责拟定写作思路,指导撰写文章并最后定稿。
