治未病脐贴的质量标准研究*
2022-05-05刘文华闵冬雨
陈 怡 刘文华 王 莉 欧 洋 韩 雪 闵冬雨
治未病脐贴是作用于神阙穴的外治药物[1],组成为:大黄、龙眼肉、花椒、艾叶以上4味,属深褐色的丸剂,有香气。大黄味苦,性寒,入胃、大肠、肝经;龙眼肉味甘,性温,归心、脾经;花椒味辛,性温,归脾、胃、肾经;艾叶味辛、苦,性温,归肝、脾、肾经。诸药苦寒甘温共济,斡旋司斯,共奏理中之效[2]。由辽宁中医药大学附属医院研发创立,应用多年临床疗效显著,为保证其质量标准,根据《中华人民共和国药典》[3](以下简称《中国药典》)各项相关规定,最终选用以大黄中的游离蒽醌和总蒽醌[4]的含量作为测定指标,用于控制本制剂质量标准,结果表明此法可用于治未病脐贴的质量控制。
1 材料与方法
1.1 仪器与试剂ZF-I型三用紫外分析仪 (上海顾村电光仪器厂),Agilent 1100液相色谱仪,Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm)。样品及对照药材、芦荟大黄素对照品、大黄酸对照品、大黄素对照品、大黄酚对照品、大黄素甲醚对照品、土大黄苷、艾叶对照药材、桉油精对照品、桂圆对照药材、花椒对照药材均来自辽宁中医药大学附属医院制剂中心。
1.2 定性鉴别
1.2.1 显微镜法取本品5 g,置离心管中,加50~60 ℃热水50 ml,振摇,使药物混悬,置离心机离心,倾去上清液,沉淀再加热水30 ml,振摇,离心,反复2次,得离心沉淀物,水浴蒸干,置显微镜下观察:外果皮细胞垂周壁连珠状增厚(花椒);T型非腺毛,顶端细胞长而弯曲,两臂不等长,柄2~4细胞,单列性非腺毛,3~5细胞,顶端细胞特长而扭曲,常断落;腺毛表面管鞋底行,由4~6细胞相对叠合而成,无柄(艾叶);草酸钙簇晶(大黄、花椒)。见图1。

注:A.非腺毛;B.腺毛;C.腺毛;D.草酸钙簇晶和内果皮细胞;E.外果皮细胞。
1.2.2 薄层色谱法(TLC)取本品3 g,剪碎,加乙酸乙酯30 ml,超声处理20 min,滤过,滤液蒸干,残渣加乙酸乙酯1 ml溶解,作为供试品溶液[5]。另取龙眼肉对照药材1 g,同法制成对照药材溶液。对照薄层色谱法(通则0502)[6]实验,吸取上述2种溶液各10 μl,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯(4∶1)为展开剂、展开、取出、晾干置紫外灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。见图2。
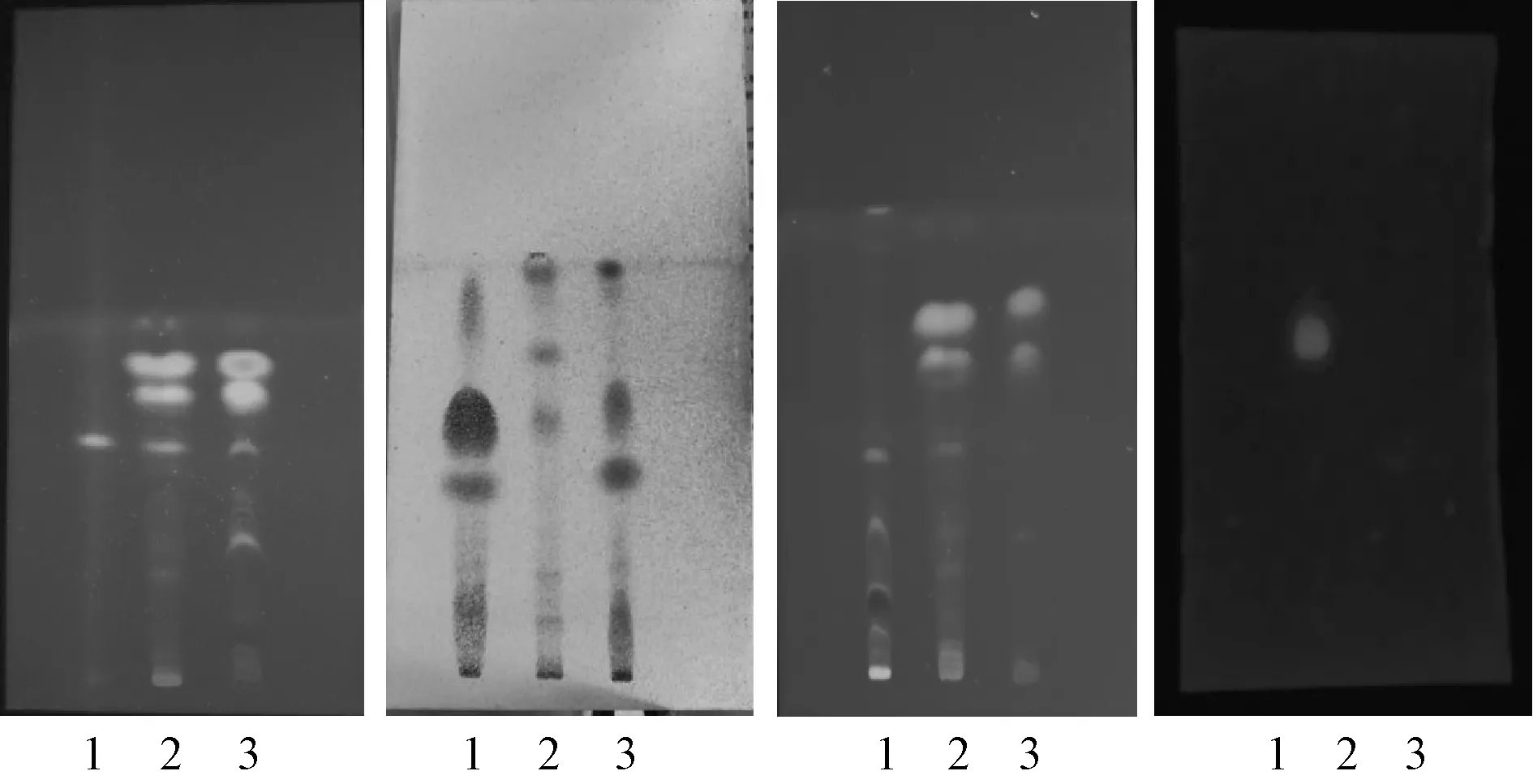
注:1.对照药材溶液;2.供试品溶液;3.阴性液。
1.3 大黄游离蒽醌的高效液相色谱含量测定方法
1.3.1 色谱条件色谱柱:Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm)。以甲醇-0.1%磷酸溶液(65∶35),检测波长254 nm。流速1.0 ml/min。柱温30 ℃。理论塔板数≥3000,分离度≥1.5,拖尾因子在0.95~1.05。
1.3.2 对照品溶液的制备精密称取芦荟大黄素对照品、大黄酸对照品、大黄素对照品、大黄酚对照品、大黄素甲醚对照品适量,加甲醇分别制成每1 ml含芦荟大黄素、大黄酸、大黄素、大黄酚各80 μg,大黄素甲醚40 μg溶液;分别精密量取上述对照品溶液各2 ml,混匀,即得(每1 ml含芦荟大黄素、大黄酸、大黄素、大黄酚各16 μg,大黄素甲醚8 μg)。见图2。
1.3.3 供试品溶液制备方法对比直接回流提取法、浸泡过夜后回流提取法和加适量硅藻土分散回流提取法后,选取含量准备、操作精当的第2种方法,取本品适量,剪碎,取约25 g,精密称定,置塞锥形瓶中,加甲醇50 ml,称定重量,放置过夜后,回流提取1 h,放冷,再称定重量,用甲醇补足减失的重量,摇匀、滤过取续滤液即得供试品溶液。
1.3.4 阴性液干扰实验取方中除大黄外的各药材,按制备工艺制成样品,再按“1.3.3供试品溶液的制备”项下制备成缺大黄阴性液。精密吸取对照品溶液、供试品溶液及阴性液各10 μl注入液相色谱仪,绘制液相色谱图,结果在与对照品色谱峰相应的位置上供试品溶液具有相同保留时间的色谱峰,阴性液在此峰位无吸收。见图2。
1.3.5 线性范围考察按上述色谱条件测定峰面积,以进样浓度为横坐标X,色谱峰面积为纵坐标Y,绘制标准曲线,经线性回归,得回归方程为:芦荟大黄素Y=47774.409X-4.429,r=0.9998,进样量在0.03125~0.5 μg内线性关系良好;大黄酸Y=43075.564X-1.258,r=0.9999,进样量在0.036375~0.582 μg内线性关系良好;大黄素Y=34449.900X-0.771,r=0.9998,进样量在0.036875~0.59 μg内线性关系良好;大黄酚Y=53092.288X-1.217,r=0.9999,进样量在0.04075~0.652 μg内线性关系良好;大黄素甲醚Y=35530.975X-6.363,r=0.9999,进样量在0.022~0.352 μg内线性关系良好。取对照品配置成不同浓度溶液,吸取10 μl,注入液相色谱仪中。见表1。

表1 线性范围考察结果
1.3.6 稳定性实验取供试品溶液,分别在0、2、4、6、8 h,按正文含量测定方法测定峰面积,结果表明,本品溶液于8 h内稳定。见表2。
表2 稳定性实验
1.3.7 精密度实验取同一对照品溶液按上述色谱条件操作,同法测定6次,结果表明,本方法精密度较高。见表3。

表3 精密度实验
1.3.8 重复性试验取同一批号样品6份,分别按样品含量测定方法测定,结果表明,本方法重复性较好。见表4。

表4 重复性实验
1.3.9 回收率试验精密称取已知含量的样品共6份,分别加入适量对照品,按供试品制备方法制备,测定含量,结果表明,本方法加样回收率良好。见表5。
表5 回收率实验
1.4 总蒽醌含量测定
1.4.1 色谱条件同[游离蒽醌]含量测定项下。
1.4.2 对照品溶液的制备取[游离蒽醌]含量测定项下对照品溶液,即得。
1.4.3 供试品溶液的制备取1.2.2项下供试品溶液,精密量取5 ml,加8%盐酸溶液10 ml,超声处理2 min,再加三氯甲烷10 ml,加热回流1 h,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取三氯甲烷层,酸液再用三氯甲烷提取3次,每次10 ml,合并三氯甲烷液,减压回收溶剂至干,残渣加甲醇使溶解,转移至10 ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
1.4.4 阴性液干扰实验取方中除大黄外的各药材,按制备工艺制成样品,再按“1.2.2供试品溶液的制备”项下制备成缺大黄阴性液。精密吸取对照品溶液、供试品溶液及阴性液各10 μl注入液相色谱仪,绘制液相色谱图,结果在与对照品色谱峰相应的位置上供试品溶液具有相同保留时间的色谱峰,阴性液在此峰位无吸收。见图2。
1.4.5 线性范围考察取对照品配置成不同浓度溶液,吸取10 μl,注入液相色谱仪中,结果见下表。
1.4.6 稳定性实验取供试品溶液,分别在0、2、4、6、8 h按正文含量测定方法测定峰面积,结果表明,本品溶液于8 h内稳定。见表6。

表6 稳定性实验
1.4.7 精密度实验同1.3.7[游离蒽醌]含量测定项下。
1.4.8 重复性实验取同一批号样品6份,分别按样品含量测定方法测定,结果表明,本方法重复性较好。见表7。
表7 重复性实验
1.4.9 回收率实验精密称取已知含量的样品共6份,分别加入适量对照品,按供试品制备方法制备,测定含量,结果表明,本方法加样回收率良好。见表8。
1.5 样品含量测定按游离蒽醌和总蒽醌含量测定方法对样品进行含量测定。见表9。

表9 样品含量测定结果
2 讨论
本品由龙眼肉、花椒、艾叶、大黄组成,根据《中国药典》[3]各项下相关规定,龙眼肉、花椒没有指标性成分可以控制含量,艾叶中以桉油精为含量测定指标,但是桉油精专属性差,因此最终选用以大黄中的游离蒽醌和总蒽醌的含量作为测定指标,用于控制本制剂质量。经过反复试验,芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚在现行色谱条件下得到了很好的分离。重复性好、精密度高、稳定性强,阴性液无干扰,在所用色谱条件下,理论塔板数≥3000,分离度≥1.5,拖尾因子在0.95~1.05,是较为成熟的含量测定方法。以此确定的质量标准测定可信度高,可以接纳为质量标准。
3 结果与结论
根据以上样品测定结果及《中国药典》[3]中大黄项下相关规定,定为本品标准为含游离蒽醌以芦荟大黄素(C15H10O5)、大黄酸(C15H8O6)、大黄素(C15H10O5)、大黄酚(C15H10O4)和大黄素甲醚(C16H12O5)的总量计,不得少于0.002%;含总蒽醌以芦荟大黄素(C15H10O5)、大黄酸(C15H8O6)、大黄素(C15H10O5)、大黄酚(C15H10O4)和大黄素甲醚(C16H12O5)的总量计,不得少于0.015%。
