基于GEO数据库挖掘与膜性肾病密切相关的关键基因
2022-04-23高燕李叶段雪婷王倩张海松李铭
高燕 李叶 段雪婷 王倩 张海松 李铭
0 引言
膜性肾病(membranous nephropathy,MN)是一种组织特异性自身免疫病,它是除糖尿病外造成成年人肾病综合征的主要病因[1-3],也是原发性肾小球肾炎中导致终末期肾脏病第二位或第三位的病因[4]。MN在各年龄段均可发病,发病高峰主要集中在50~60岁[5]。MN的病理改变主要表现为肾小球基底膜不断增厚,在电镜下可以观察到上皮下区域内出现电子致密沉积物,足细胞足突融合[6-7]。所以发现IgG抗体颗粒样沉积于肾小球毛细血管袢是MN的诊断依据[8]。临床上患者主要表现为肾病综合征或者无症状蛋白尿,蛋白尿量每天超过3.5 g,约有1/3的患者可以自愈,1/3的患者会发展为终末期肾病,另有1/3的患者表现为持续存在蛋白尿但肾功能仍可长期维持[1]。
自2002年以来与MN有关的自身抗原包括中性内肽酶(neutral endopeptidase)[9]、M型磷脂酶A2受体(muscle-type phospholipase A2 receptor,PLA2R)[10],以及2014年报道的1型血小板反应蛋白结构域7A(thrombospondin type-1 domain-containing 7A,THSD7A)[11]不断被鉴定出来,为MN的诊断带来了革命性的变革。但是这些自身抗原的发现并没有揭示MN的发病机制,所以目前自身抗原对MN的形成发展机制尚不明确,因而在临床上使用昂贵且具有潜在毒性的药物针对抗原治疗MN仍具有很大的挑战性,也给患者和社会带来沉重的经济负担。
近年来兴起的基因芯片以及生物信息学研究技术为揭示肾脏疾病的病例机制提供了新的研究策略。Rudnicki等[12]通过基因芯片结合生物信息学分析揭示了进行性肾病的microRNA以及mRNA的表达谱改变,Ju等[13]则通过对肾组织进行转录组学研究发现表皮生长因子(epidermal growth factor,EGF)可以作为慢性肾病进展的标记物。Garantziotis等[14]则通过生物信息学方法挖掘出一些新的与包括MN在内的原发性肾小球病有关的药物靶点。差异表达的miR-150-5p则被鉴定出来可以预测IgA肾病的进展情况[15]。由此可以看出转录组学结合生物信息学方法已成为深入研究肾脏相关疾病发病机制的有力工具。因此本研究依托GEO(Gene Expression Omnibus)数据库中MN的相关表达芯片数据,通过差异基因表达分析,基因功能注释、富集分析以及蛋白互作分析,最终获得与MN密切相关的基因以及相关信号通路,为以后揭示MN的分子病理机制提供理论研究基础。
1 材料和方法
1.1 试验材料
在GEO数据库检索获得数据集GSE108113,该数据集以GPL19983 [HuGene-2_1-st] Affymetrix Human Gene 2.1 ST Array [HuGene21st_Hs_ENTREZG_19.0.0] 为平台,包含280个样本,其中包含正常肾小球组织6例,正常肾小管组织5例,MN患者肾小球组织44例,MN患者肾小管组织43例,局灶性和节段性肾小球硬化患者肾小球和肾小管组织共76例,微小病变肾小球和肾小管组织34例,ANCA相关性小血管炎肾小球和肾小管组织共72例。从中提取获得6例正常肾小球样本和44例MN肾小球样本的数据。
1.2 基因表达分析
首先对获得的50个样本进行层次聚类分析,检测样本之间的相关性,剔除离群样本之后对数据集进行主成分分析(principal component analysis,PCA)。之后通过R包org.Hs.eg.db将芯片探针转换为基因名后,再用R包limma进行分析获取在正常肾小球组织和MN肾小球组织之间差异表达的基因,设置筛选条件为P<0.01,且log2FC>2 或 log2FC<-2,并通过R语言对差异表达基因绘制火山图和热图进行展示。
1.3 基因功能注释以及富集分析
通过R包clusterProfiler[16]对获得的差异表达基因进行GO(gene ontology)功能注释分析,设置阈值P<0.01,q<0.01,差异具有统计学意义。同样的方法也进行了KEGG(Kyoto Encyclopedia of Genes and Genomes)信号通路分析富集,P<0.05,差异具有统计学意义。
1.4 筛选核心基因
将获得的差异表达基因导入STRING(https://string-db.org/)在线数据库,绘制蛋白互作网络图,设置阈值maximum number of interactors=0,confidence score ≥ 0.4,并通过Cytoscape软件进行可视化,运用Cytoscape中的MCODE(molecular complex detection)插件对整个蛋白互作网络进行聚类分析,筛选得分最高的功能模块,将其节点作为核心基因。
2 结果与分析
2.1 筛选MN组织与正常组织之间的差异表达基因
从GEO数据库下载获得数据集GSE108113,通过R软件从中提取6例正常肾小球组织的数据以及44例MN肾小球组织的数据,首先对这50例样本进行聚类分析,结果发现离群样本GSM2889924[图1(a)],将其剔除之后再进行PCA分析,两组样本聚类结果一致[图1(b)]。之后运用R软件里的limma包对两组样本进行差异表达基因分析,通过筛选标准|log2FC|>2,P<0.01,共获得上调差异表达基因36条,下调差异表达基因126条。最后对获得的差异表达基因绘制火山图以及热图展示其在两组样本中的分布,如图1(c)和图1(d)所示。

图1 筛选与MN有关的差异表达基因Figure 1 Identification of differentially expressed genes in association with MN
2.2 GO分析
运用R软件里的clusterProfiler包对获取的差异表达基因进行GO分析,结果如图2所示。发现在“分子功能”(molecular function,MF)方面,差异表达基因主要富集在珠蛋白结合、亚铁血红素结合、氧气结合、四吡咯结合以及氧载体活性[图2(a)];差异表达基因在“生物过程”(biological process,BP)方面主要分布在药物分解代谢以及血小板脱颗粒[图2(b)];差异表达基因富集到的“细胞组分”(cellular component,CC)主要包括血液微粒(blood microparticle),细胞顶端(apical part of cell)及其质膜(apical plasma membrane),囊泡(vesicle lumen)以及胞质囊泡(cytoplasmic vesicle lumen)等[图2(c)]。
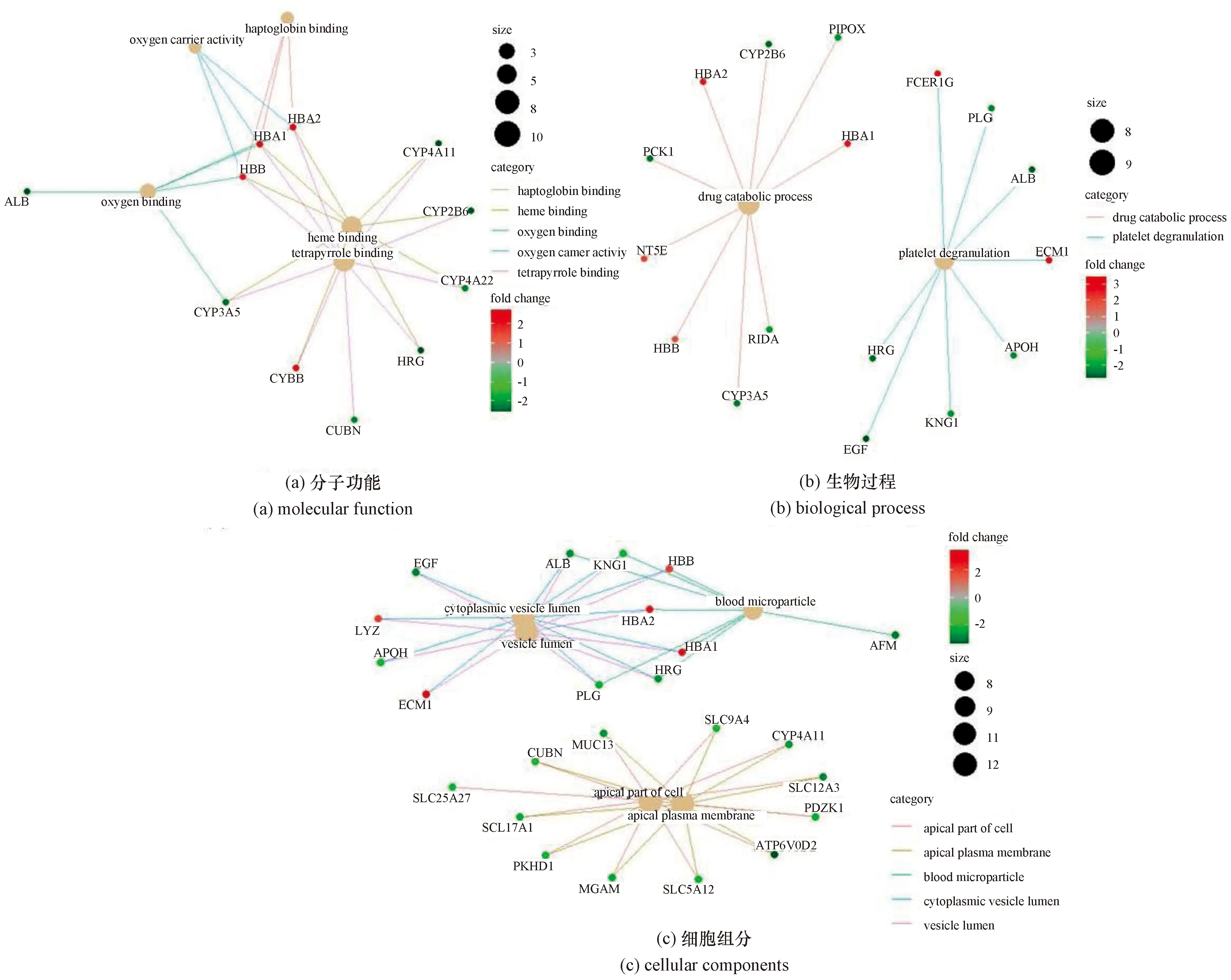
图2 GO条目及其所包含的差异表达基因Figure 2 Including genes of the GO terms
2.3 KEGG通路分析
同样通过R软件里的clusterProfiler包对筛选出来的差异表达基因进行KEGG通路富集分析,结果如图3所示,结果中红色代表上调的差异表达基因富集的通路,其主要富集在吞噬体、疟疾以及非洲锥虫;蓝色代表下调的差异表达基因富集的通路,其主要富集的信号通路包括细胞色素P450参与的药物代谢、视黄醇代谢、PPAR信号通路以及糖代谢、脂肪代谢等。
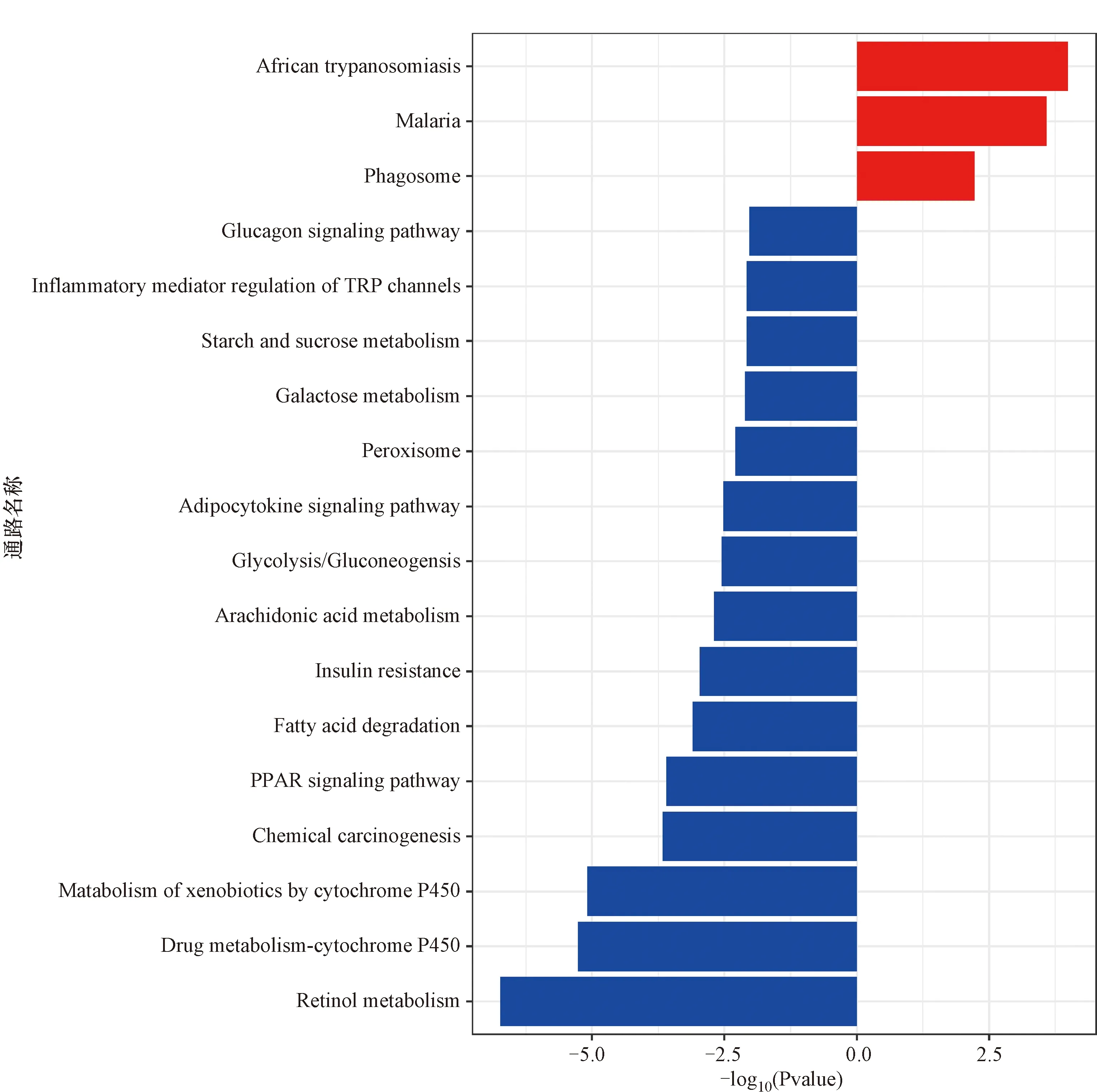
红色代表上调的差异表达基因富集的通路,蓝色代表下调的差异表达基因富集的通路。图3 KEGG富集分析的结果Figure 3 KEGG enrichment analysis of differentially expressed genes
2.4 蛋白互作分析
将获得的162个差异表达基因输入在线网站STRING构建蛋白互作网络图,之后将获得结果导入Cytoscape软件进行可视化以及后续分析。最终获得由60个节点117条边构成的蛋白互作网络复合体,其余的102个差异表达基因未被包含到蛋白互作网络复合体中[图4(a)]。接下来通过Cytoscape中的MCODE插件对所构建的整个蛋白互作网络进行聚类关联分析,设置参数degree cutoff=2,node score cutoff=0.2,k-core=2 以及max. depth=100,并选择分值最高的Cluster作为最终筛选出来的核心基因,该Cluster包含13个节点25条边,得分为4.167[图4(b)],由此共得到13个核心基因(表1)。
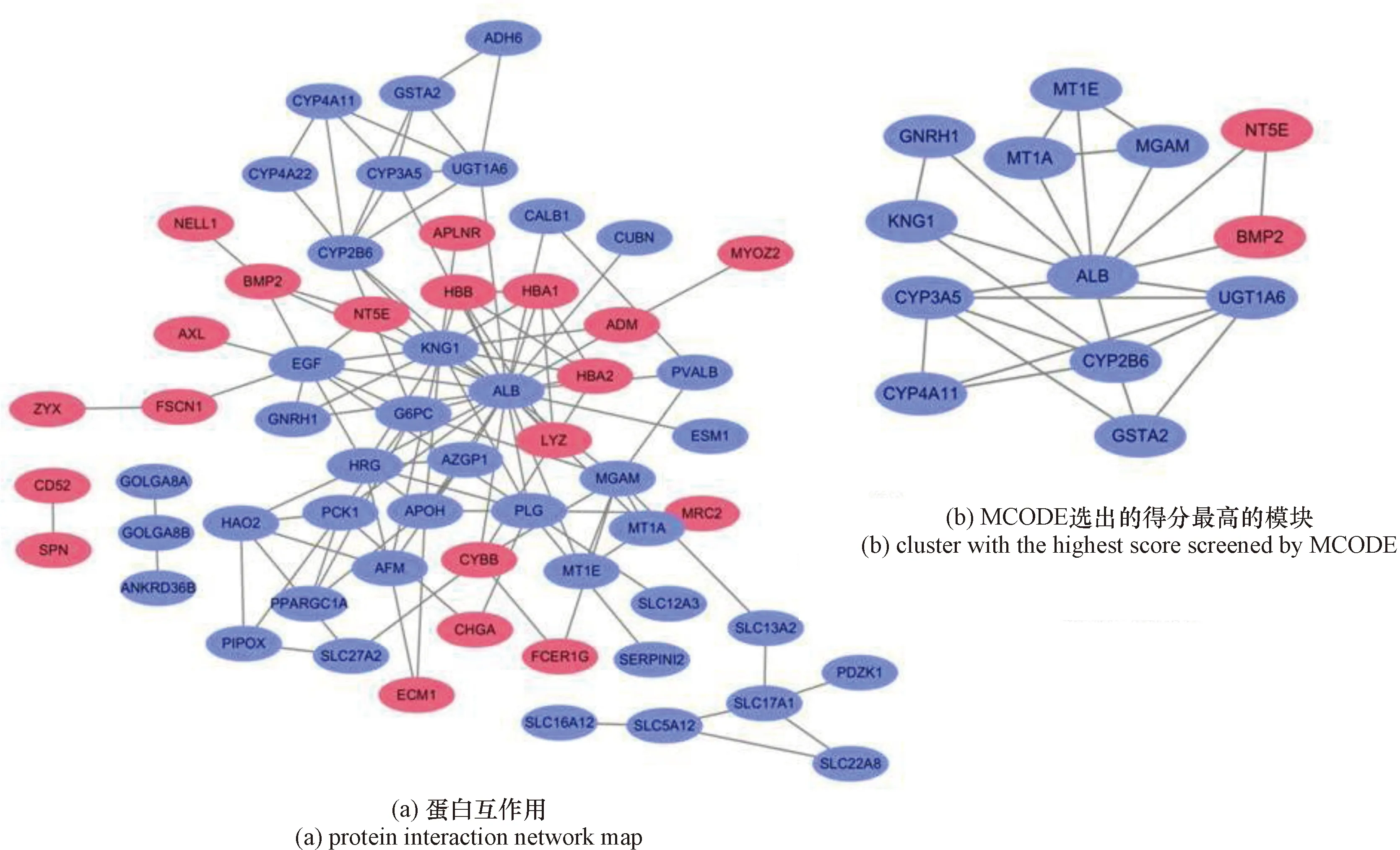
图4 蛋白互作复合体Figure 4 PPI network

表1 与MN有关的核心基因Table 1 The core genes related with MN
3 讨论与结论
MN自然病程长,是一种慢性病,而近年来在我国PM2.5污染严重的城市MN的发病率升高明显,已成为继IgA肾病之后第二大原发性肾小球病[17],给社会经济以及人们的日常生活带来沉重负担。因而揭示MN的发病机制,寻找有效的治疗靶点成为急需解决的医学问题。本研究通过生物信息学方法提取并分析了GEO数据库的数据集GSE108113中关于MN的数据,最终获得与MN密切相关的13个核心基因以及药物代谢、视黄醇代谢等相关信号通路。
CYP3A5(cytochrome P450 family 3 subfamily A member 5),CYP4A11(cytochrome P450 family 4 subfamily A member 11),CYP2B6(cytochrome P450 family 2 subfamily B member 6)均属于细胞色素P450家族,这3个基因广泛参与到药物代谢过程,在本研究中还富集到了视黄醇代谢过程。Déri等[18]也报道了在终末期肾病患者的肾组织里面细胞色素P450家族的基因表达下调,与本研究的变化趋势一致,说明了细胞色素P450家族在肾病发生过程中发挥着重要作用。另外被富集到这2个通路的核心基因还有GSTA2(glutathione S-transferase alpha 2)和UGT1A6(UDP glucuronosyltransferase family 1 member A6)。GSTA2属于谷胱甘肽-S-转移酶家族,是II期解毒酶家族。Kim等[19]在研究中发现非马沙坦防治肾脏纤维化可能是通过上调GSTA2以降低肾脏的氧化损伤,因而高水平表达的GSTA2对于肾脏具有保护作用。UGT1A6则属于UDP-葡萄糖醛酸基转移酶1家族的一员,它可以将激素、药物等降解为水溶性的排泄物。Liu等[20]发现肝脏及肾脏的葡萄苷酸化作用很大程度上受到该家族基因表达的影响。因而UGT1A6表达降低可能会影响肾脏的正常功能。
其他的核心基因像NT5E(5′-nucleotidase ecto)是一种核苷酸酶,也参与药物的代谢。而Cappelli等[21]也观察到TGF-β可以激活肾小管上皮细胞表达NT5E以促进肾脏纤维化,而在糖尿病肾病患者尿液中NT5E含量升高可以作为预后不良的指标。激肽原1(KNG1,kininogen 1)在GO分析中被注释到血液微粒、血小板脱颗粒以及胞质囊泡中。Tang等[22]通过生物信息学方法发现KNG1与糖尿病患者的肾小管损伤有关。
本研究揭示了以CYP3A5、CYP4A11、CYP2B6
为代表的13个核心基因表达异常,以及药物代谢和视黄醇代谢通路与MN之间存在的密切关联。然而这些基因表达异常与MN发展形成的病理分子机制还有待深入研究。同时本研究中只选取一个数据集中的部分合适样本,研究有一定局限性,后续还需要有更多的实验数据验证支持。本研究将为阐释MN发生的病理机制提供新的研究视角和研究基础。
