Molecular docking study of xylogranatins binding to glycogen synthase kinase-3β
2022-04-19ChristianBaillyrardVergoten
Christian Bailly,Gérard Vergoten
a.Scientific Consulting Office,OncoWitan,Wasquehal,Lille 59290,France
b.University of Lille,Inserm,INFINITE - U1286,Institut de Chimie Pharmaceutique Albert Lespagnol(ICPAL),Faculty of Pharmacy,3 rue du Professeur Laguesse,BP-83,F-59006,Lille,France
ABSTRACT Objective The mangrove tree Xylocarpus granatum J.Koenig(X.granatum)is a medicinal plant used to treat various diseases in several Asian countries.Many bioactive natural products have been isolated from the plants,particularly several groups of limonoids,including 18 xylogranatins(Xyl-A to R),all of which bear a furyl-δ-lactone core commonly found in limonoids.Based on a structural analogy with the limonoids obacunone and gedunin,we hypothesized that xylogranatins could target the enzyme glycogen synthase kinase-3β(GSK-3β),a major target for the treatment of neurodegenerative pathologies,viral infections,and cancers.Methods We investigated the binding of the 18 xylogranatins to GSK-3β using molecular docking in comparison with two known reference GSK-3β ATP-competitive inhibitors,LY2090314 and AR-A014418.For each compound bound to GSK-3β,the empirical energy of interaction(ΔE)was calculated and compared to that obtained with known GSK-3β inhibitors and limonoid triterpenes that target this enzyme.Results Five compounds were identified as potential GSK-3β binders,Xyl-A,-C,-J,-N,and-O,for which the calculated empirical ΔE was equivalent to that calculated using the best reference molecule AR-A014418.The best ligand is Xyl-C,which is known to have marked anticancer properties.Binding of Xyl-C to the ATP-binding pocket of GSK-3β positions the furyl-δlactone unit deep into the binding-site cavity.Other xylogranatin derivatives bearing a central pyridine ring or a compact polycyclic structure are much less adapted for GSK-3β binding.Structure-binding relationships are discussed.Conclusion GSK-3β may contribute to the anticancer effects of X.granatum extract.This study paves the way for the identification of other furyl-δ-lactone-containing limonoids as GSK-3β modulators.
Keywords Natural products Xylocarpus granatum Xylogranatins Glycogen synthase kinase-3β(GSK-3β)Limonoids Cancer Molecular modelling Structure-activity relationship
1 Introduction
The mangrove treeXylocarpus granatumJ.Koenig(X.granatum)is used in traditional medicine in different Asian countries to treat symptoms,such as fever,dysentery,and diarrhea,associated with diseases such as malaria and cholera[1,2].Extracts prepared from various parts of the plant,mostly the fruits,leaves,and bark,are used as folk medicines in India,Bangladesh,and other countries to reduce fever or treat diarrhea[3].These medicinal activities are linked to the presence of a large number of bioactive secondary metabolites in the extracts[4].The plant is a rich reservoir of active natural products,including flavonoids,alkaloids,terpenoids,and steroids[5].Many compounds with antioxidant,anti-inflammatory,antiparasitic,antidiabetic,and anticancer properties have been identified inX.granatumextracts in recent years[6].Limonoids are particularly abundant and diverse in this plant,including xylomexicanins A - N isolated from the leaves and twigs[7],hainanxylogranins A - X found in the leaves and bark[8],granatripodins A and B from the seeds[9],thaigranatins A - T,and krishnagranatins A - I isolated from the seeds[10-12]and many other related limononoids.An important series of limonoids found inX.granatumare named xylogranatins A - R,which are essentially isolated from the fruits[13-16]and seeds[17]of the Chinese mangrove.These represent a homogeneous series of 18 limonoids(Figure 1).The lead product in this series is xylogranatin C(Xyl-C),which has anticancer properties[18].Xyl-C suppresses tumor cell proliferationin vitroand exerts marked pro-apoptotic activities[18].The analog Xyl-B also exhibits anticancer effects,with significant cytotoxicity against colon cancer cell lines[19].However,the mechanisms of action of various xylogranatins remain unknown.Therefore,this study focused on identifying potential molecular targets for these natural products.
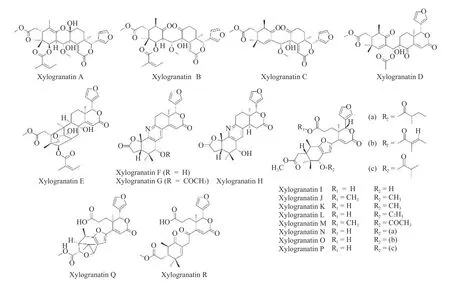
Figure 1 Structures of the xylogranatins
Certain limonoid triterpenes can modulate the activity of the serine/threonine kinase glycogen synthase kinase-3β(GSK-3β),which regulates a wide range of cellular processes(Figure 2).This is the case for the limonoid triterpene obacunone isolated from the root bark ofDictamnus dasycarpusTurcz[20]and the structurally related limonoid gedunin isolated from the plantAzadirachta indica,which also functions as an effective GSK-3βinhibitor[21].Obacunone is an antioxidant,anti-inflammatory,and anticancer agent found in diverse plants[22,23],whereas gedunin is an inhibitor of melanogenesis and an inducer of apoptosis[24,25].The compounds obacunone and gedunin possess a common furyl-δ-lactone core,which is also present in xylogranatins.This analogy prompted us to investigate the potential capacity of xylogranatins to bind GSK-3βusing molecular docking.Several crystal structures of GSK-3βare available in the protein data bank.We selected structure 1Q5K,which corresponds to GSK-3βbound to the anticancer inhibitor AR-A014418.This well-resolved three-dimensional(3D)structure(1.94 Å)corresponds to the activated form of human GSK-3βbound to AR-A014418 in its ATP pocket[26].We evaluated the capacity of each of the 18 xylogranatins to form stable complexes with GSK-3βand discovered that five compounds(Xyl-A,-C,-J,-N,and -O)could potentially be considered as bona fide GSK-3βligands,with the capacity to form stable complexes comparable to those of the two reference compounds tested in parallel:the GSK-3βATP-competitive inhibitors LY2090314 and AR-A014418.
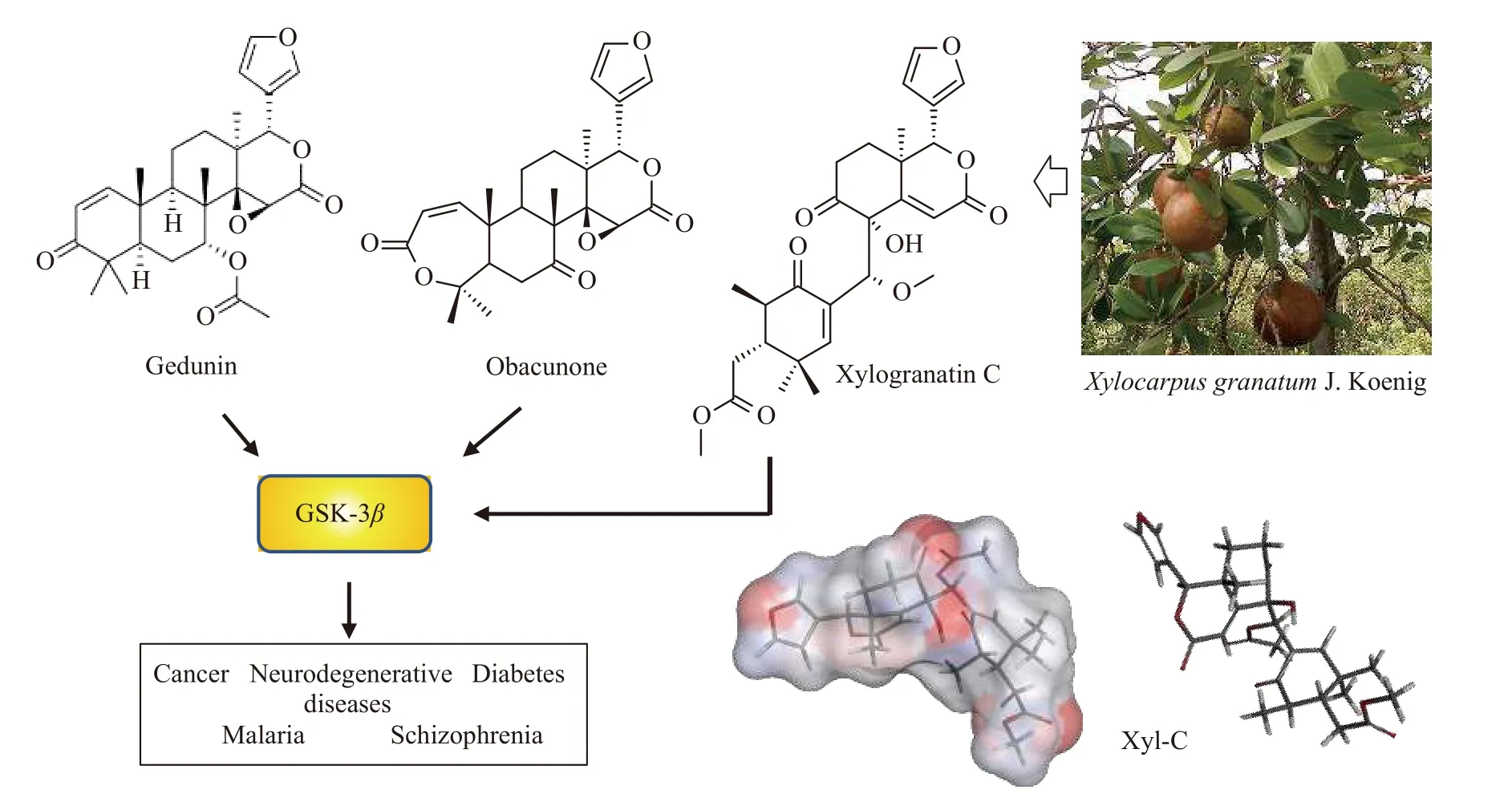
Figure 2 Structures of gedunin and obacunone,known to interfere with GSK-3β,an enzyme implicated in a diversity of diseases
2 Materials and methods
2.1 Molecular structures and software
3D structures of GSK-3βin complex with a small-molecule inhibitor(AR-A014418)were retrieved from the Protein Data Bank(PDB)(www.rcsb.org)under PDB code 1Q5K[26].Docking experiments were performed using the GOLD software(GOLD 5.3 release,Cambridge Crystallographic Data Centre,Cambridge,UK).Before starting the docking procedure,the structure of the ligands was optimized using a classical Monte Carlo conformational search procedure,as described in the Biochemical and Organic Simulation System(BOSS)software,freely available to academic users[27].Potential GSK-3β-binding sites for the different products were searched using the web server CASTp 3.0(Computed Atlas of Surface Topography of proteins)and visualized with the molecular modeling system Chimera 1.15[28,29].
2.2 In silico molecular docking procedure
With each structure,based on the shape complementarity criteria,the drug-binding site,and surrounding amino acids were defined.Shape complementarity and geometry considerations favor a docking grid centered in the volume defined by the central amino acid.Within the binding site,the side chains of the specific amino acids were considered fully flexible.For structure 1Q5K,the flexible amino acids are Lys85,Val110,Leu132,Asp133,Tyr134,Val135,Glu137,Arg141,Leu188,and Cys199.The ligand was defined as flexible during docking.Up to 100 poses that were energetically reasonable were retained while searching for the correct binding mode of the ligand.The decision to maintain a trial pose is based on ranked poses,using the Piecewise Linear Potential(PLP)fitness scoring function(which is the default in GOLD version 5.3 used here)[30].The same procedure was used to establish molecular models for the various natural products listed in Table 1 and reference compounds.The empirical potential energy of the interactionΔE for the ranked complexes was evaluated using the simple expressionΔE(interaction)=E(complex)-[E(protein)+ E(ligand)].Therefore,the spectroscopic empirical potential energy function Spectroscopic Potential Algorithm for Simulating Biomolecular Conformational Adaptability(SPASIBA)and its corresponding parameters were used.The free energies of hydration(ΔG)were estimated using the molecular mechanics/generalized born surface area(MM/GBSA)model in Monte Carlo simulations using BOSS software[31].The stability of the receptor-ligand complex was evaluated using the empirical potential energy of interaction[32,33].The MM/GBSA procedure was used to evaluate the free energies of hydration(within the BOSS program)[27]in relation to aqueous solubility.Molecular graphics and analyses were performed using Discovery Studio Visualizer,Biovia 2020(Dassault Systèmes BIOVIA Discovery Studio Visualizer 2020;San Diego,Dassault Systèmes,2020).
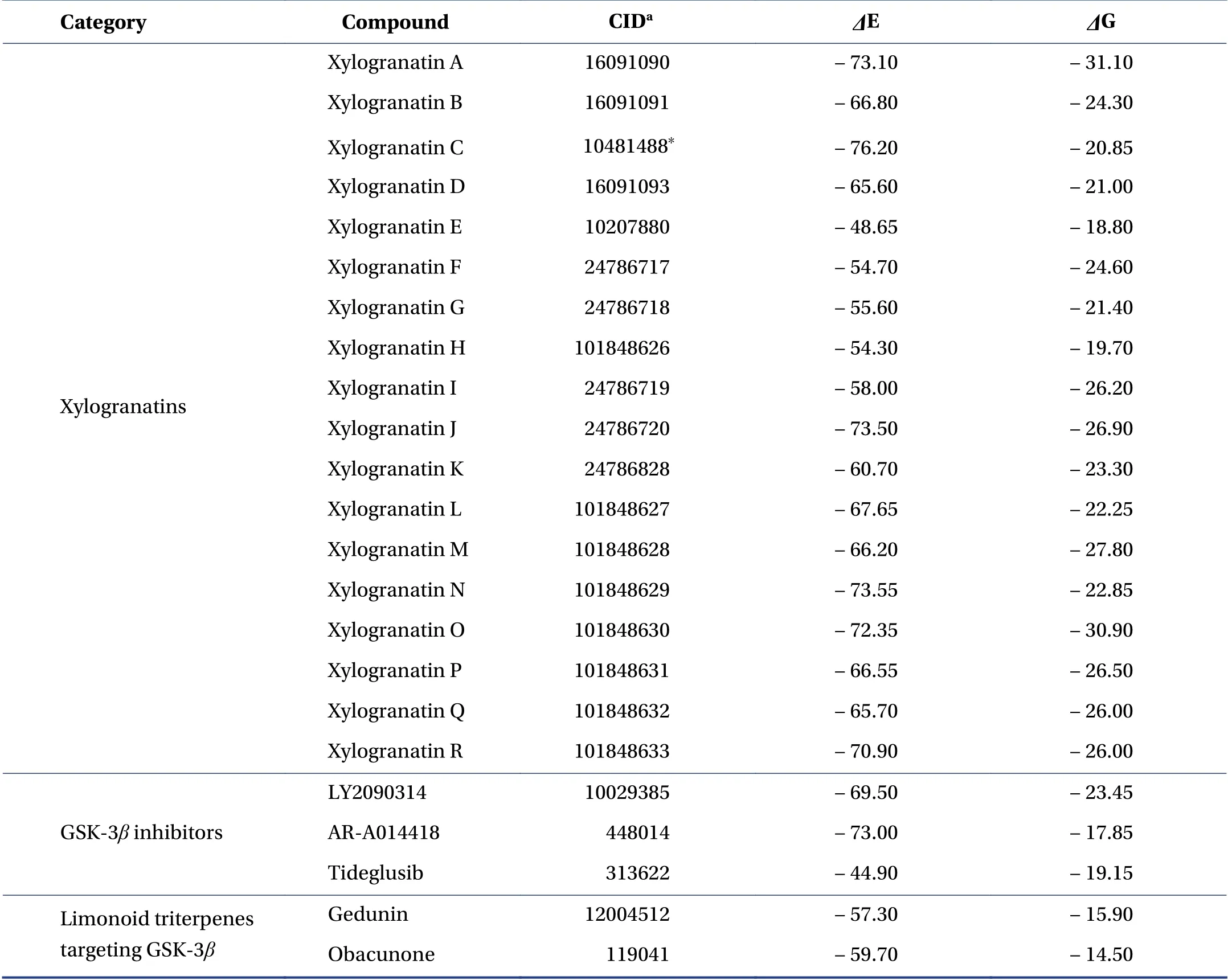
Table 1 Calculated potential energy of interaction(ΔE,kcal/mol)and free energy of hydration(ΔG,kcal/mol)for the interaction of xylogranatins with the GSK-3β protein(PDB:1Q5K)
2.3 Monte Carlo simulations
Molecular dynamics(MD)and Monte Carlo(MC)simulations are the two most widely used methods for investigating protein-ligand stability and affinity.MD simulation of proteins is a challenge that requires carefulconsideration of the 1st law of thermodynamics[34].Both methods use an empirical force field to control the total energy(MC,energy minimization)and forces(MD,Newton’s equations of motion).To use MD simulations confidently,a force field parameterized for dynamic properties is required.The development of a reliable and accurate force field for conformational analysis is important.This requires the accuracy of the force field over the entire potential surface rather than in the region of the global minimum[35].The most commonly used academic force fields(CHARMM,AMBER,and GROMOS)do not exhibit the required vibrational spectroscopic quality.Minimizing a protein structure with normal modes can result in the calculation of imaginary wavenumbers corresponding to maxima in the potential energy(transition states,mainly owing to inadequate barriers to internal rotation).The spectroscopic SPASIBA force field has been specifically developed to provide refined empirical molecular mechanics force field parameters,as described in other studies[32,36].Therefore,we used MC simulations rather than MD simulations,which require a substantial increase in computer time to achieve the same level of convergence[37].
3 Results
3.1 Molecular models of reference compounds bound to GSK-3β
The docking analysis was performed with the two limonoid triterpenes,obacunone and gedunin,which have been previously shown to inhibit GSK-3β.Their propensity to form stable complexes with the enzyme was compared to that of the reference compounds,tideglusib,LY2090314,and AR-A014418.For each compound,we calculated the empirical energy of interaction(ΔE)and energy of hydration(ΔG).In all cases,the ligand-binding site corresponded to the site found in the crystal structure.For LY2090314 and AR-A014418,the predicted docking poses in the ligand-protein complex were excellent(ΔE=-69.5 and-73.0 kcal/mol,respectively),whereas the docking score was much less favorable with tideglusib(ΔE=-44.9 kcal/mol).This is expected because this compound has been shown to bind the inactive(DFG-out)conformation of GSK-3β[38].This analysis aimed to identify natural products with GSK-3βbinding capacity at least equivalent or superior to those of LY2090314 and AR-A014418.
3.2 Molecular models of xylogranatins bound to GSK-3β
Obacunone and gedunin can form equally stable complexes with GSK-3β,withΔE values in the range -57 to-60 kcal/mol,that is,approximately 20% less than the values calculated with the two synthetic references(Table 1).Thereafter,we tested each of the 18 xylogranatins compounds bearing a furyl-δ-lactone core.The calculatedΔE values ranged from -48.6 kcal/mol for Xyl-E to-76.2 kcal/mol for Xyl-C,respectively the less adapted and the best suited compound for binding to GSK-3β.Compounds were categorized into three groups.The calculatedΔE values were >- 60 kcal/mol for Xyl-E,-F,-G,-H,and -I,suggesting that these five compounds are not well adapted to interact with GSK-3β.ΔE values comprised between -70 and -60 kcal/mol were measured for Xyl-B,-D,-K,-L,-M,-P,and -Q,respectively.Five compounds emerged from the analysis,withΔE values <
- 70 kcal/mol:the lead compound Xyl-C and the analogs Xyl-A,-J,-N,and -O.For these top five compounds,the formation of a stable complex with GSK-3βcan be considered because theΔE values are comparable to those measured with the best reference,AR-A014418.
3.3 Configuration of the Xyl-C-GSK-3β complex
A molecular model of Xyl-C binding to GSK-3βis shown in Figure 3.This natural product binds to the ATP active site of the enzyme,which is deeply enfolded by two domains.The furyl-δ-lactone unit of Xyl-C points toward the interior of the site,whereas the acetyl group is oriented toward the external side of the cavity.This cavity is relatively large and accommodates a variety of molecules.The extended conformation of Xyl-C was well-adapted to fit into the pocket.In contrast,more rigid molecules,such as Xyl-E,-F,-G,and -H,which present a tetracyclic core,are much less suited to sit in the binding cavity.Compounds with a pyridine ring,such as Xyl-F,-G,and -H,do not offer a suitable skeleton for GSK-3βbinding.However,the structure-binding relationships are complex because Xyl-A,also equipped with a rigid skeleton,presents a good GSK-3βbinding affinity.The only common element among all molecules is the furyl-δ-lactone core,which is directly implicated in protein interactions.In the case of Xyl-C,the furan ring interacted with the protein via Hbonds with residues Asp-200 and Lys-85(Figure 4).This situation is slightly different from that of Xyl-N,which is another good binder,with the implication of Lys-85 and a π-sulfur interaction with Cys-199.For compounds such as Xyl-N,-O,and -A,the orientation in the binding pocket is slightly different;however,in all cases,there are key residues implicated in the H-bonds,such as Lys-85 and Asp-200,in addition to a range of van der Walls and alkyl contacts.The same situation was observed with Xyl-J,one of the top 5 GSK-3βbinders in our series.Here again,the compound inserts its furyl-δ-lactone unit into the site deep into the hydrophobic cavity(Figure 5).Xyl-I,-J,-K,and -L differed by the single carbon atom at R1/R2.Structure-binding relationships are complex;however,it appears that a methyl ester at position R2is preferred over an acid function.The nature of the R2substituent is less important and a bulky group is tolerated.The tigloyl or isobutyryl R2side chain of Xyl-O and -P protruded outside the cavity,with little interaction with the protein.
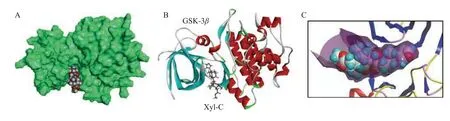
Figure 3 Molecular model of Xyl-C bound to GSK-3β(PDB:1Q5K)
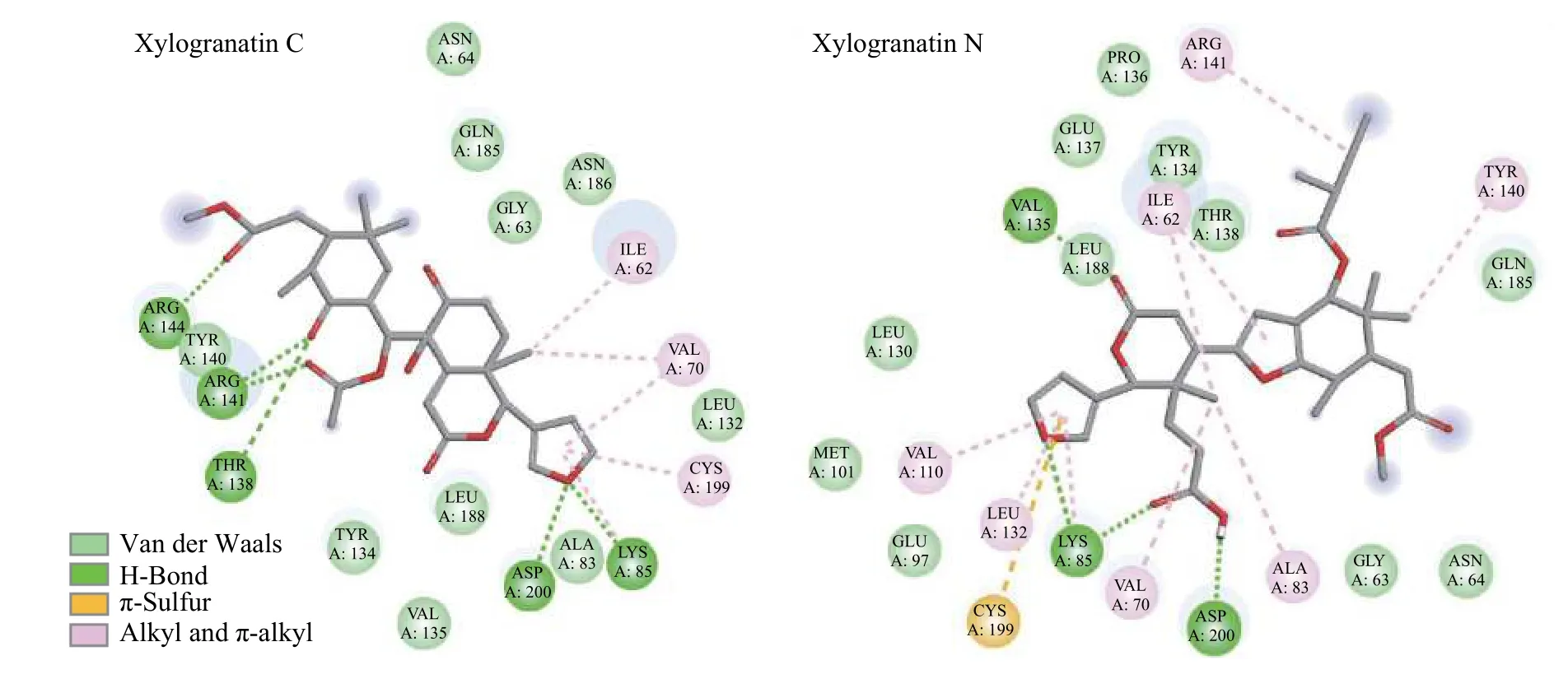
Figure 4 Binding map contacts for Xyl-C and -N bound to GSK-3β

Figure 5 Close-up view of Xyl-J bound to GSK-3β
4 Discussion
Xylogranatins A - R represent a small group of limonoids isolated from the fruits or seeds of the mangrove treeXylocarpus granatumJ.Koenig,used in traditional medicine[13-17].These natural products display anti-inflammatory effects,and at least two members of the series,xylogranatins B and C,have anticancer properties[18,19].Xyl-C was recently found to significantly suppress tumor cell proliferation and trigger apoptosis[18].However,the molecular targets of these compounds are currently unknown.Based on the analogy with other limonoids,such as gedunin and obacunone bearing a furyl-δ-lactone unit,we hypothesized that xylogranatins might interact with the enzyme GSK-3β,a serine/threonine kinase involved in a large range of cellular processes.This hypothesis was tested using molecular docking with the crystal structure of GSK-3β.
The discovery that GSK-3βcould contribute to the pharmacological action of certain xylogranatin derivatives is significant because it provides a potential molecular basis to explain the anticancer effects ofX.granatumextracts and Xyl-C[18,39].GSK-3βis considered a promising target for overcoming chemoresistance,particularly in pancreatic cancer[40,41].Overexpression of GSK-3βplays a direct role in tumor growth and modulates drug response[42,43].Therefore,it is important to identify novel GSK-3βinhibitors that can regulate the activity of this key enzyme.The discovery of new GSK-3βbinders such as Xyl-C could impact the design or development of novel therapeutics.Small molecules bearing a furyl-δ-lactone unit could be designed to target this enzyme.GSK-3βhas also been implicated in neurological disorders,multiple sclerosis,and diabetes[44-46].The targeting of GSK-3βwith xylogranatins may also partly explain the antidiabetic action ofX.granatumextracts[47].Natural and synthetic scaffolds that modulate GSK-3βactivity have been used to target glucose metabolism[48].This study provides new ideas for the design of GSK-3βligands based on furyl-δ-lactone units.
Among the 18 xylogranatin derivatives tested,five compounds revealed a binding capacity comparable to that of the two synthetic products LY2090314 and ARA014418,which were used as reference inhibitors.The potential capacity of Xyl-A,-C,-J,-N,and -O to form stable complexes with GSK-3βwas superior to that measured with the two limonoid products at the origin of our analysis.The measuredΔE values(- 73/- 76 kcal/mol)were similar to those of the best-known binder,ARA014418.Therefore,we hypothesized that GSK-3βrepresents a potential protein target for these compounds,particularly the tumor-active product Xyl-C.We are aware of the limitations of molecular docking evaluation;however,it is a useful approach to guide the discovery of a target for these molecules with no mechanism of action defined at present.
Many compounds are structurally affiliated with xylogranatins,such as xylogranatopyridines A - B and xylogranatumines A - G fromX.granatum[49],and other limonoids with a furyl-δ-lactone core.Synthetic approaches are also being developed,offering opportunities to design related limonoids[50-52].All of these compounds could be further tested as potential inhibitors of GSK-3β.Considering the implication of this enzyme in many vital biological processes,validation of ourin silicoproposal is eagerly awaited.Thus,Xyl-C may be a very useful GSK-3βmodulator.
5 Conclusion
The molecular docking study reported in this study identified five xylogranatin derivatives(Xyl-A,-C,-J,-N,and-O)as potential GSK-3βbinders.We compared the GSK-3βbinding capacity of all 18 xylogranatins identified thus far from the medicinal plantXylocarpus granatumJ.Koenig.Xyl-C is revealed as a potential GSK-3βligand,and its furyl-δ-lactone unit is directly implicated in protein interactions.Four other xylogranatins from the mangrove tree were considered GSK-3βbinders:xylogranatins A,J,N,and O.GSK-3βcould contribute to the antidiabetic and anticancer effects reported previously with extracts ofX.granatum.This discovery will orient future studies towards a more detailed analysis of the contribution of GSK-3βto the pharmacological effects of xylogranatins,notably the anticancer effects of Xyl-C.These observations will help to better understand and rationalize the antidiabetic effects ofX.granatumextract.
6 Highlights
(i)Xylogranatins A - R are limonoids isolated from the medicinal plantXylocarpus granatumJ.Koenig.
(ii)Molecular docking revealed that xylogranatins A,C,J,N,and O can bind to GSK-3β.
(iii)Structure-binding studies indicated a key role for the furyl-δ-lactone core of the products.
(iv)GSK-3βlikely contributes to the neuroprotective and anticancer activities of Xyl-C.
Competing interests
The authors declare no conflict of interest.
