CoMFA Study on Anti-proliferative Activity of Fluoroquinolone Amide Derivatives①
2022-04-16FENGHuiCAOJingPeiFENGChangJun
FENG Hui CAO Jing-Pei FENG Chang-Jun
a (School of Chemical Engineering and Technology, China University of Mining and Technology, Xuzhou 221116, China)
b (Key Laboratory of Coal Processing and Efficient Utilization (Ministry of Education), Xuzhou 221116, China)
c (School of Material & Chemical Engineering, Xuzhou University of Technology, Xuzhou 221018, China)
ABSTRACT The in vitro anti-proliferative activity (pICi, i = hp, ca, hl) of fluoroquinolone (rhodanine α,β-unsaturated ketone)amide compounds, referred to as “fluoroquinolone amide derivatives (FQADs)” towards Hep-3B, Capan-1 and HL60 cells, was studied by the 3D-QSAR method of comparative molecular field analysis (CoMFA). Based on the training set of 14 compounds,the prediction model was established, which was further verified by the test set of 5 compounds with template molecule included.It is found that steric and electrostatic fields contribute 66.8% and 33.2% to pIChp, 61.4% and 38.6% to pICca, and 61.5% and 38.5% to pIChl, respectively. The Rcv2 (i.e, cross-validation coefficient) is 0.324, 0.381, and 0.421 for pIChp, pICca, and pIChl,respectively, while the corresponding R2 (i.e, non-cross-validation coefficient) all reach 0.999. Then, the models were employed to estimate the activities of the training and test compounds, and the results show that the stability and predictability of developed models are very satisfactory. According to the contour maps of steric and electronic fields, bulky groups linked to 2-, 3-,4-positions of phenyl ring, and electropositive groups near the 4-position and electronegative groups far away may increase the anti-proliferative activity. Using the information provided by the 3D contour maps, four new FQADs owing higher antiproliferative activity were designed, but their effectiveness should be further tested by experiments.
Keywords: fluoroquinolone amide derivatives, Hep-3B cell, Capan-1cell, HL60 cell, anti-proliferative activity, comparative molecular field analysis; DOI: 10.14102/j.cnki.0254-5861.2011-3343
1 INTRODUCTION
Due to their extensive pharmacological activities, fivemembered heterocyclic ketones especially rhodanine have become the dominant skeleton of medicinal chemistry and have been widely used in the construction of molecular chemical skeleton of new drugs. As for fluoroquinolone, the carboxyl group of C-3 site is not a necessary pharmacophore for anti-proliferative activity. However, if the C-3 amido group is used as the connecting chain to carry another pharmacophore with anti-proliferative activity, the combination of different pharmacophores and the superposition of activities can be realized, and as a result, its antibacterial activity can be effectively converted into anti-proliferative activity[1,2].Gaoet al.[3]spliced the rhodanineα,β-unsaturated ketone skeleton with anti-proliferative fluoroquinolone skeleton using amido as the connecting chain to synthesize 18 fluoroquinolone (rhodanineα,β-unsaturated ketone) amide compounds, referred to as “fluoroquinolone amide derivatives,FQADs”. Then, they used MTT method to determine the in vitro anti-proliferative activities of these compounds towards human hepatoma (Hep-3B) cells, human pancreatic cancer(Capan-1) cells and human leukemia (HL60) cells, which were expressed asIC50.hp,IC50.caandIC50.hl, respectively or as a general term ofIC50.i, in the unit ofμmol·dm-3.
The early research methods of quantitative structure-activity relationship (QSAR) mainly involve two-dimensional structural information of molecules, which are known as 2D-QSAR[4-10]. At present, the three-dimensional structure properties of molecules are introduced into QSAR to develop“3D-QSAR” models[11-17]. Therefore, compared with traditional 2D-QSAR, 3D-QSAR methods can reflect the real image of the interaction between bioactive molecules and receptors more accurately, and reveal the mechanism of drugreceptor interaction more deeply. Furthermore, the fitting results of 3D-QSAR are generally better than 2D-QSAR.Recently, the comparative molecular field analysis (CoFMA)method[11-17]has been increasingly used in 3D-QSAR researches, because it not only contains clear physical and chemical information, but also indirectly reflects the nonbonding interaction between molecules and targets.
In this paper, the 3D-QSAR model for anti-proliferative activity (pICi)[3]of FQADs was established by CoMFA method. An intuitive structure-activity isoelectric diagram was obtained to reveal the molecular structure factors affecting pICiand explore the microscopic mechanism of antiproliferative activity, which would provide theoretical reference for designing new and highly-efficient FQAD antiproliferative drugs.
2 MATERIALS AND METHODS
At present, computer-aided drug design can be divided into receptor-based and ligand-based methods. Since the crystal structure of FQADs receptor has not been analyzed yet, the ligand-based method was used to construct CoMFA model.
2. 1 Biological activity data of studied compounds
The parent structure of 18 FQADs synthesized by Gaoet al.[3]is presented in Fig. 1, with the corresponding substituents Ar shown below.
TheIC50.i(i= hp, ca, hl, mol·dm-3) determined by MTT method is shown in Table 1. According to the basic principles of physical chemistry, a logarithmic relationship exists between the change of reaction free energy and the equilibrium concentration of components[18]. Therefore, pICi,i.e., the negative logarithm ofIC50.i(pICi= -logIC50.i), was used in CoMFA model, and the specific values are listed in Table
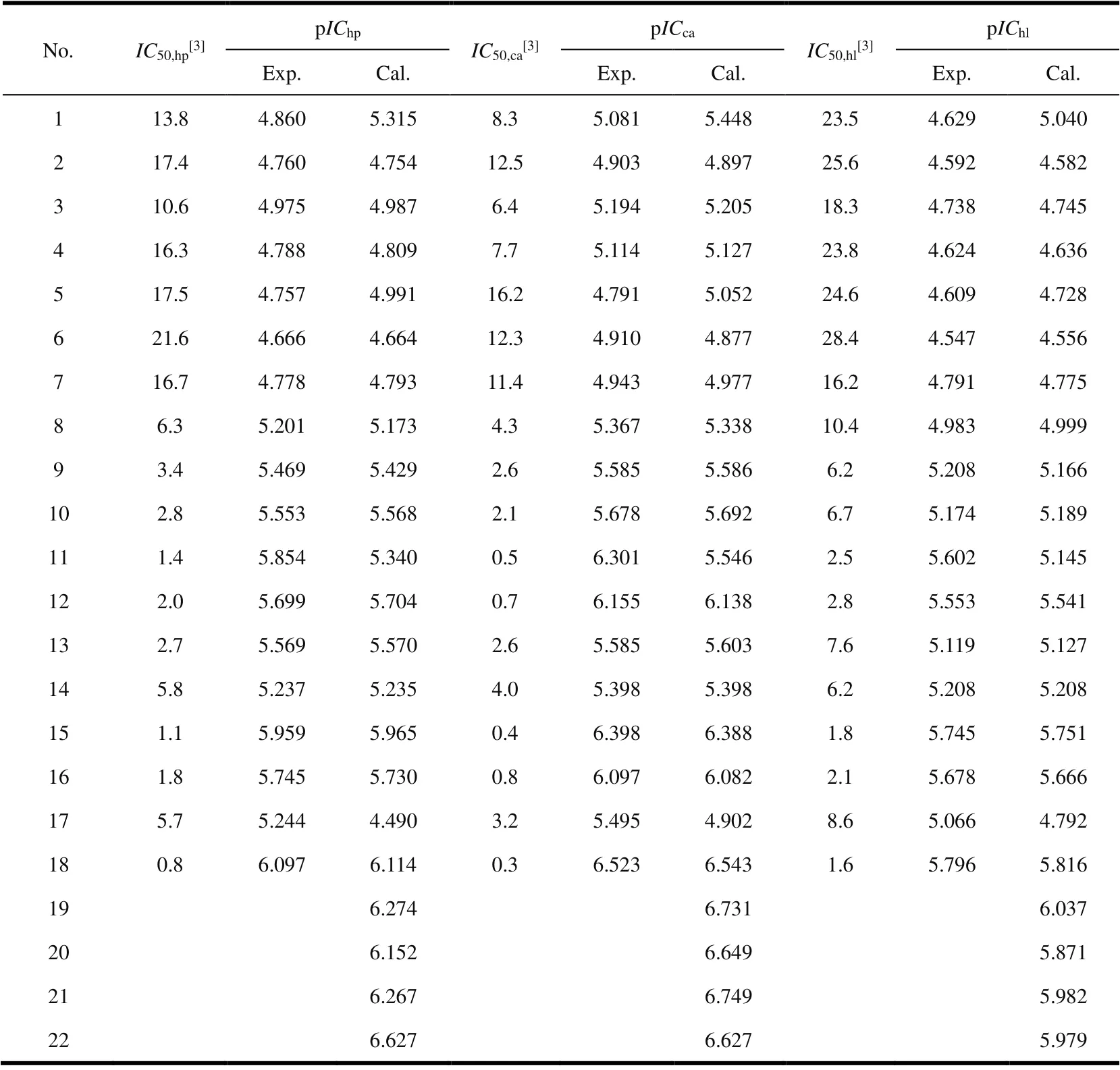
Table 1. Comparison of Experimental and Calculated Anti-proliferative Activities (pICi) of FQADs
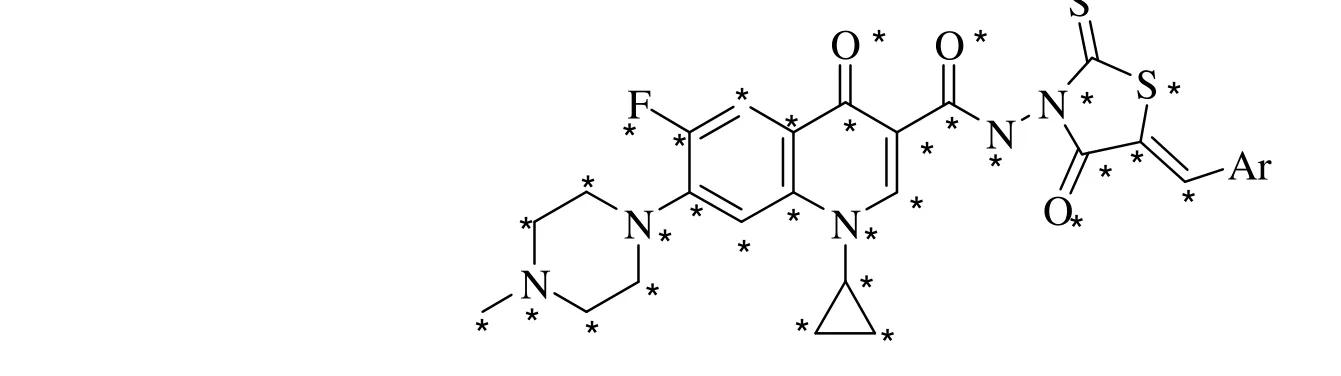
Fig. 1. Basic structure of FQADs
2. 2 Minimization and alignment
In this paper, Sybyl-x2.1.1, the latest molecular simulation software of Tripos Company, was used to establish CoMFA model of in vitro anti-proliferation activity (pICi) of FQADs on human Hep-3B, Capan-1 and HL60 cells. The key to modeling is to establish the bioactive (pharmacodynamic) conformation of drug molecules and to superpose compound molecules according to reasonable matching rules.
In this study, the molecular conformation with the lowest energy was used instead of the bioactive conformation(pharmacodynamic conformation). Molecular structures were sketched by the sketch module in Sybyl-x2.1.1 software,which were then minimized with Tripos force field using the Gasteiger-Huckel charges and conjugated gradient method.Unless otherwise stated, all default parameters were used in CoMFA. As one of the most sensitive processes in CoMFA analysis, structural alignment directly determines the CoMFA model’s prediction accuracy and the contour maps’ reliability.Molecule 18 that has the most potent activity was used as a template molecule for alignment. DATABASE ALIGNMENT command in Sybyl was used to align all homologues to the template molecule by rotation and translation, so as to minimize RMSD (i.e., root-mean-square error) between atoms in the template and the homologue. The alignment results based on above strategy are shown in Fig. 2. Subsequently, these alignments were used to calculate probe interaction energy in CoMFA.
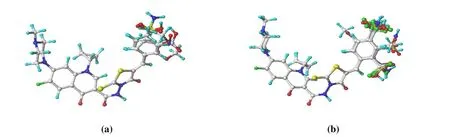
Fig. 2. 3D view of all aligned molecules for the training set (a) and test set (b)
2. 3 Model generation and validation
CoMFA model for biological activity (pICi)[3]of FQADs was established by 14 superposed training set molecules. The Steric (St) and Electrostatic fields (El) on each grid node around the superposed molecules were calculated using Tripos standard force field. The threshold (Cut off value) of field energy was set to 125.5 kJ⋅mol-1, and the default values were adopted for the other parameters.
PLS (i.e., partial least squares) analysis was performed after the input of pICiinto the dataset of the training set. First,the cross-validation correlation coefficient (Rcv2) and the optimal number (N) of component were determined by leaveone-out (LOO) cross-validation method. WhenRcv2> 0.3[19],the established model is statistically significant at the significance level of 5%, and its chance correlation probability is less than 5%, that is, the reliability of the model is 95%. Then,the robustness and statistical confidence of the 3D-QSAR models derived was further assessed by non-cross-validation correlation coefficient (R2) using non-cross-validation method. It is generally believed that the model withR2> 0.9[20]gives a good fitting result, because it reveals structural factors that affect more than 90% of the biological activity. In addition, the statistical variance ratio (F) is also a test index to measure the significance level of QSAR model. Finally, the view of CoMFA module is adopted to provide the threedimensional equipotential maps of stereo field and static electric field to directly reflect their contributions to biological activity.
3 RESULTS AND DISCUSSION
3. 1 3D-QSAR model
Based on CoMFA analysis, 3D-QSAR models of FQADs were established, which are given in Table 2 with the corresponding statistical parameters.
HighR2(> 0.9[20]) andRcv2(> 0.3[19]) as well as smallSDare obtained, indicating that the CoMFA models established are reliable and predictive. At the significance level of 99%,theFcritical value (F0.01(6, 8)) is only 6.37. TheFvalues of the three models are much larger than 6.37, indicating that they are all statistically significant. Moreover, the contribution of steric field is greater than that of electrostatic field(61~67%vs.33~39%). Therefore, the steric interaction of the ligand is a more important factor affecting the antiproliferation activity.
The anti-proliferative activity of the training set molecules is estimated by the established CoMFA models. It can be seen from Table 2 that the calculated values are largely consistent with the corresponding experimental values. In addition, the external test set was used to test the 3D-QSAR model’s external prediction ability. Estimated values of anti-proliferative activities of the four compounds (see “pICi.cal.” of No. 1, 5, 11 and 17 in Table 1) in the test set are very close to the corresponding experimental data, and therefore the 3D-QSAR models have good external prediction ability.
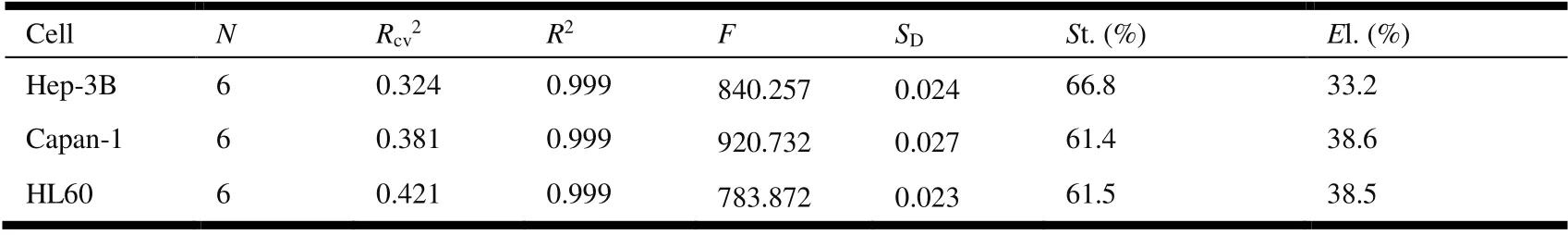
Table 2. Statistical Parameters of the CoMFA Models of the Training Set Molecules
3. 2 Analysis of the three-dimensional equipotential diagram
Based on the equipotential diagram of force field coefficients obtained by CoMFA model, the important regions related to the activity are determined in three-dimensional steric and electrostatic fields to effectively explain the molecular structure characteristics of active compounds and the underlying molecular mechanisms of action.
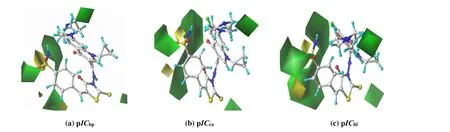
Fig. 3. CoMFA steric contour maps with compound 18 as the template molecule:(a) pIChp; (b) pICca; (c) pIChl. Green and yellow color denotes sterically favored and disfavored areas, respectively
3. 2. 1 Steric contour map of pICi
The stereogram of CoMFA model usually includes two regions, where the introduction of a large substituent in the green region or a small group in the yellow region is helpful to improve the activity of the compound.
Fig. 3 gives the CoMFA steric contour map of pICi, with the most active compound 18 being the template molecule.The contour maps of pIChp, pICcaand pIChlare very similar.First, there are two large green regions above the 4-position of the benzene ring, and thus introducing large-volume groups herein will enhance the anti-proliferative activity. For example, compounds 9, 15, 17 and 18 have larger pICi, because there is a larger substituent at the 4-position of benzene ring. Secondly, small yellow regions are observed at the 3-and 4-positions, suggesting that the presence of small-volume groups is beneficial to the improvement of anti-proliferative activity. For instance, pICivalues of compounds 10 and 16 are smaller than those of compounds 9 and 15, respectively.
3. 2. 2 Electrostatic contour map of pICi
As for the contour map of electrostatic field, blue and red contours denote the regions that bioactivity will be increased by the presence of electron-donating and electron-withdrawing groups, respectively.
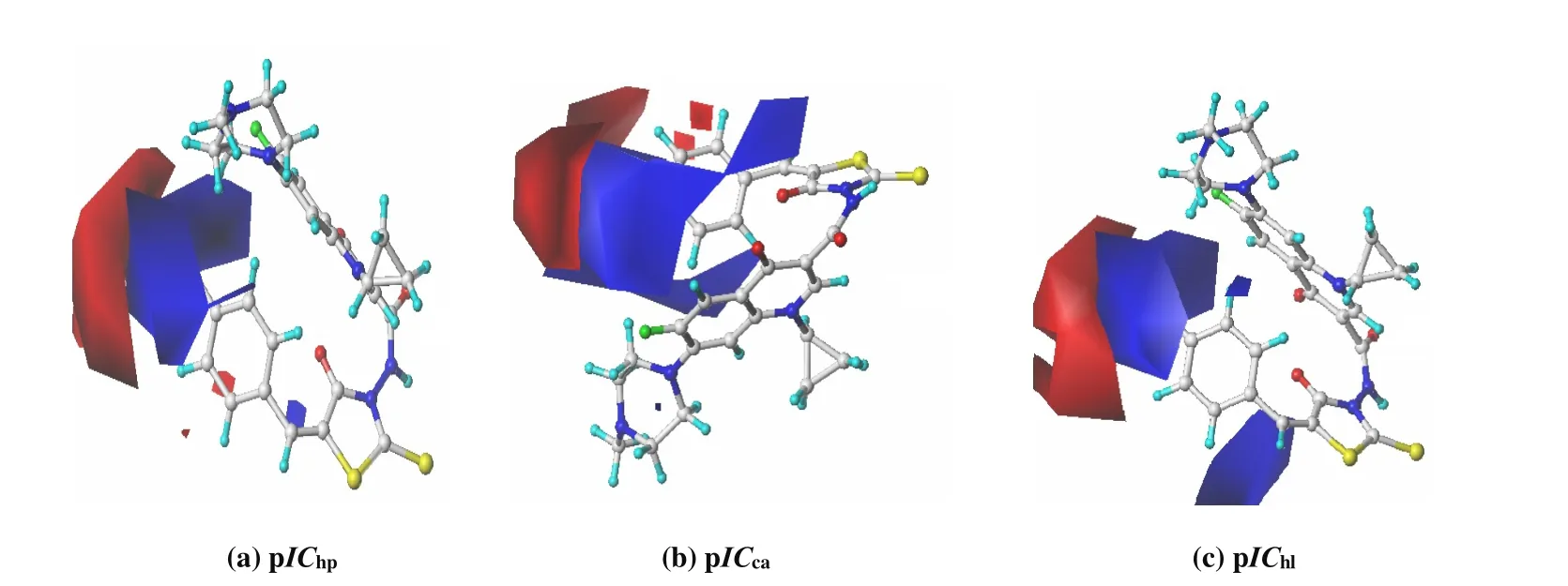
Fig. 4. Contour maps of CoMFA electrostatic field with compound 18 as the template molecule: (a) pIChp; (b) pICca; (c) pIChl.The favorable sites for positively and negatively charged groups are depicted by steric blue/red polyhedra
Fig. 4 shows the CoMFA electrostatic contour map of pICi,with the most active compound 18 as the template molecule.As seen from Fig. 4, the contour maps of pIChp, pICcaand pIChlare very similar, with a big blue area near the 4-site of the benzene ring and a red area far away. The electrostatic distribution of this shape coincides with the charge distribution of -NO2, -CO2H, -SO2NH2and other groups, due to the following two reasons: The N, C and S atoms with small electronegativity are attached to the benzene ring at the 4-position, and the positive electronegativity is just in the blue region; Meanwhile, these atoms are bonded to the more electronegative O and N atoms, which are negatively charged in the red region. Therefore, the pICiof compounds 9, 10, 15,16 and 18 containing -NO2, -CO2H and -SO2NH2groups are all larger.
The 3D-QSAR model of the training set gives the contribution of stereoscopic and electrostatic fields to pICi(Table 2), which shows that stereoscopic effect has greater influence on anti-proliferative activity than the electrostatic one. Accordingly, the anti-proliferative mechanism of FQADs involves the hydrophobic interaction between FQADs and biological enzymes in cancer cells, the steric hindrance between groups attached to common skeleton and cavities in enzymes,and the associated Coulomb force and hydrogen bonding, the effects of which decrease in turn. The three-dimensional equipotential diagrams of the three CoMFA models are very similar, which indicates that FQADs have similar mechanisms for inhibiting the proliferation of human Hep-3B, Capan-1 and HL60 cells in vitro.
3. 3 Theoretical design of new FQADs with higher bioactivity
One of the purposes of QSAR research is to design molecules according to the model and predict the activity of designed molecules with the model, so as to provide a theoretical basis for the design of new highly active molecules. It can be seen from the 3D-equipotential diagram in section 3.2 that the existence of bulky groups at the 2-, 3- and 4-sites of benzene ring and the presence of a positively charged central group near the 4-position and a negatively charged group far away will improve the in vitro inhibitory activity of FQADs.Based on this rule, four compounds (the last four in Table 1)were designed by introducing groups such as -CO2H and-SO2NH2at the 2-, 3- and 4-sites of benzene ring. The pICi.cal.values of these four compounds estimated by the CoMFA model are larger than that of the template molecule 18, which indicates that they have stronger anti-proliferative activity than the template molecule. Thus, these four compounds are expected to be developed as new anti-proliferative drugs.Interestingly, the predicted value of No. 20 is lower than that of No. 19, because there is a large group attached to the 3-position (yellow area) of No. 20 molecule. Similar phenomenon was also observed in the No. 22 compound when comparing with the No. 21 one. However, No. 22 has a larger value than No. 21 in terms of pIChp, because the yellow area of 3-position is very small. Despite the large pICi.cal.values,the anti-proliferative activities of these compounds need to be further verified by clinical medical experiments.
4 CONCLUSION
From this study, the following conclusions can be reached:
(1) Comparative molecular force field method (CoMFA)was used to investigate the three-dimensional quantitative structure-activity relationship of in vitro anti-proliferative activity (pICi) of FQADs on human hepatoma (Hep-3B)cells, human pancreatic cancer (Capan-1) cells and human leukemia (HL60) cells, and three 3D-QSAR models with good correlation, stability and prediction ability were established.
(2) The CoMFA model well explained the anti-proliferative mechanism of FQADs, which involved hydrophobic interaction, steric hindrance of substituent groups, Coulomb force and hydrogen bonding between FQADs and biological enzymes in cancer cells in a decreasing order of effect.
(3) According to the similarity of three-dimensional equipotential diagrams of three CoMFA models, it is speculated that FQADs have similar inhibition mechanisms on the proliferation of human hepatoma Hep-3B, Capan-1 and HL60 cells in vitro are similar.
(4) The four designed compounds have stronger antiproliferative activity than the template molecules, and are expected to be developed as new anti-proliferative drugs of the same kind.
杂志排行

结构化学的其它文章
- Structural and Electronic Properties of Lutetium Doped Germanium Clusters LuGen(+/0/-) (n = 6~19):A Density Functional Theory Investigation①
- Discovery of Benzimidazole Derivatives as Novel Aldosterone Synthase Inhibitors: QSAR, Docking Studies, and Molecular Dynamics Simulation①
- QSAR Models for Predicting Additive and Synergistic Toxicities of Binary Pesticide Mixtures on Scenedesmus Obliquus①
- Preparation, Crystal Structure and Fungicidal Activity of N-(5-(benzofuranol-7-oxymethyl)-1,3,4-thiadiazol-2-yl)amide Compounds①
- Antibiotic Silver Particles Coated Graphene Oxide/polyurethane Nanocomposites Foams and Its Mechanical Properties①
- Planar Tetracoordinate Carbon in 6σ + 2π Double Aromatic CBe42- Derivatives①