动力学水合物抑制剂性能与官能团作用研究进展及展望
2022-04-13王佳琪张昕宇贺佳乐葛坤
王佳琪,张昕宇,贺佳乐,葛坤
(哈尔滨工程大学动力与能源工程学院,黑龙江哈尔滨,150000)
天然气水合物又称“可燃冰”,是由水和烃类小分子气体甲烷、乙烷等在高压、低温条件下形成的笼型结构的晶体物质,又被称为笼型水合物[1]。天然气广泛存在于永久冻土和深海等自然环境中,早在1924年,HAMMERSCHMIDT[2]发现在天然气输送管道中存在天然气水合物的二次生成。近年来,油气开采从陆地转向海洋,在深海水合物勘探、天然气试生产过程中,深海较高的静水压力与较低的环境温度为水合物的形成创造了有利条件,造成油气开采井筒、运输管线的堵塞与损坏[3],减少油气产量与运输量,造成巨大经济损失,阻碍天然气产业发展[4]。因此,有效控制水合物的形成、防止水合物堵塞、保障运输管线流动安全是促进天然气行业发展的重要因素。目前,水合物二次生成预防与流动保障有几种不同方法,包括降低压力、使用隔热管道、加热管线以及这几种方法的结合,其中,经济性更好的化学试剂法得到广泛应用。
水合物抑制剂主要包括传统热力学水合物抑制剂(THIs)和低剂量水合物抑制剂(LDHIs)。热力学抑制剂能够改变天然气水合物生成的热力学条件,使其平衡温度降低,平衡压力升高,从而起到抑制水合物生成的作用。但其使用剂量大(需达到一定质量分数才能达到理想抑制效果)、难以回收以及循环利用,使用成本较高,并且部分热力学抑制剂还具有一定毒性。低剂量水合物抑制剂(LDHIs)包括动力学水合物抑制剂(KHIs)及防聚剂(AAs),具有低量、高效等优点。其中,防聚集(AAs)通常是一些聚合物或表面活性剂,用于油水两相体系,其并不能抑制水合物的生成,但可以通过与液态烃的相互作用防止水合物聚集结块,从而抑制水合物的进一步形成,保证管线的正常流动。虽然其用量较少,但应用场合有限,若处于无油、无水环境则无法发挥抑制水合物生长的作用,并且防聚剂需均匀分散在流体中才能发挥其最大抑制效果。另外,其价格较贵,通常与KHIs和THIs结合使用。近年来,经济环保、适用范围广的动力学水合物抑制剂(KHIs)受到广泛关注。动力学水合物抑制剂(KHIs)通常是一类水溶性聚合物[5],动力学抑制剂通过自身官能团的作用抑制水合物成核或者阻碍晶核的生长,其使用剂量很低,但抑制效果显著,可使用成本约为热力学抑制剂的50%[6]。研究表明,动力学抑制剂分子的官能团对动力学抑制剂的性能起到至关重要的作用,并且基于目前对各类官能团的认识,对动力学抑制剂进行设计、改性研究成为当前研究热点。本文作者对相应官能团抑制机理以及改性得到的新型抑制剂对水合物的抑制行为及其性能、影响因素等进行总结,并提出未来动力学抑制剂的发展趋势。
1 水合物形成与动力学抑制剂作用机理
1.1 水合物成核机理假说
一个完整的水合物生成过程一般包含气体溶解、成核(晶核的形成)和生长(晶核生长成晶体)3个阶段[1]。
1.1.1 簇成核假说
SLOAN 等[7]在1991年提出了不稳定簇假说,CHRISTIANSEN等[8]修正了该假设,认为水合物成核过程首先从纯水分子开始进行。该假说认为在没有客体分子的纯水当中存在短暂、不稳定的五元环或六元环结构。随着客体分子溶解,环绕在客体分子周围的不稳定环状结构周围将立即形成不稳定簇。不稳定的团簇逐渐生长、结合并聚集成临界尺寸的核,随后生长。
1.1.2 局部结构成核假说
RADHAKRISHNAN 等[9]提出局部结构成核假说,随后,MOON 等[10]基于甲烷水合物的分子动力学模拟也提出类似假说。局部结构成核假说认为由于热量波动,部分溶解的客体分子将有序排列成与水合物相类似的空间构型,当有序排列的客体分子的数量超过临界核中客体分子的数量时,其周围的水分子结构将会受到扰动并与有序排列的客体分子相互作用,形成临界尺寸的核。
1.1.3 界面成核假说
LONG 等[11-12]提出了界面成核假说。与不稳定团簇和局部结构成核假说不同,该假说认为成核是从气水界面的气相一侧开始的。气体分子被运移并吸附于气液界面,随后通过表面扩散,运移至水分子事先形成的半笼状孔穴,而后形成部分或完全的笼,持续这个过程,更多的气体和水分子加入到形成的空穴中,直到临界核形成并生长。
1.1.4 混合团簇(blob hypothesis)成核假说
JACOBSON 等[13]提出了混合团簇假说(blob hypothesis)。该假说认为水合物结晶过程为溶液→形成混合团簇(blob)→无定形包合物→水合物晶体。具体过程如下。
第一步:过饱和溶液可逆地形成混合团簇。混合团簇是由水分子和被水分子分隔的多个客体分子形成的聚集体,其与周围的水分子相互作用,在质量和热量波动的情况下,混合团簇中笼状体重复生成和分解,直到一个笼状体团簇达到临界尺寸为止。
第二步:当一个笼状体团簇达到临界尺寸时,通过共享笼状体的各个面,临界尺寸团簇开始进行空间填充式的晶体增长过程,并通过共享新连接的笼子的表面而变成无定形的笼形物。无定形笼形物是一种水合物前体的亚稳定中间体,因为它的混合结构合成了几种瞬态的笼形结构,这些结构的笼形和所占总数比例与水合物最终晶体结构中的笼形和所占总数比例不同。无定形笼形物与混合团簇的不同之处在于:在混合团簇中,水分子还没有被锁定在笼中,而在无定形笼形物中,水分子通过氢键作用锁定在笼结构上。
第三步:稳定的sI 和亚稳定的sII 水合物的晶体在第二部的无定形包合物的连续生长和成熟中形成。
在某种程度上,混合团簇假说是不稳定簇和局部结构假说的结合[13],它强调水分子和气体分子的作用以及它们在形成和稳定团簇结构中的相互作用。
1.2 水合物的生长机理
经过成核阶段后,生长立即开始,随着大量气体溶解,生长颗粒聚集。VYSNIAUSKAS等[14-15]认为水合物的生长受界面面积、温度、压力的影响。ENGLEZOS等[16]认为晶体生长过程分为2个阶段:溶解气体从液态扩散到晶体与液相界面上的扩散阶段;界面处气体分子与周围水分子结合形成稳定笼形结构的阶段。UCHIDA等[17-18]通过拉曼光谱技术研究了气体组成、温度、压力对水合物类型的影响并分析了二氧化碳水合物的生长过程,认为二氧化碳水合物的形成需要大量二氧化碳的溶解。根据前人对水合物生长的研究成果,水合物生长过程受水合物晶体表面的生长速率、水和客体分子向生长表面的传质过程、水合物晶体生长过程中热量的释放传递的共同作用。
1.3 基于水合物成核的动力学抑制剂作用机理
1.3.1 吸附抑制机理
抑制剂分子通过电负性强的官能团与水合物晶体表面的水分子形成氢键吸附在水合物晶体表面,占据水合物的生长位点从而抑制笼型的生长,其他官能团则在空间上阻碍晶体表面的生长,吸附抑制机理示意图如图1所示[19]。但抑制剂的吸附过程十分复杂,YAGASAKI 等[20]使用PVCap 单体研究了KHIs的抑制机理,通过对比PVCap单体从水移动到水合物表面前后的自由能发现,酰胺基团与水合物表面水分子间的氢键并不是聚合物分子吸附作用的驱动力所致,而是水合物空穴引起的熵稳定所致。
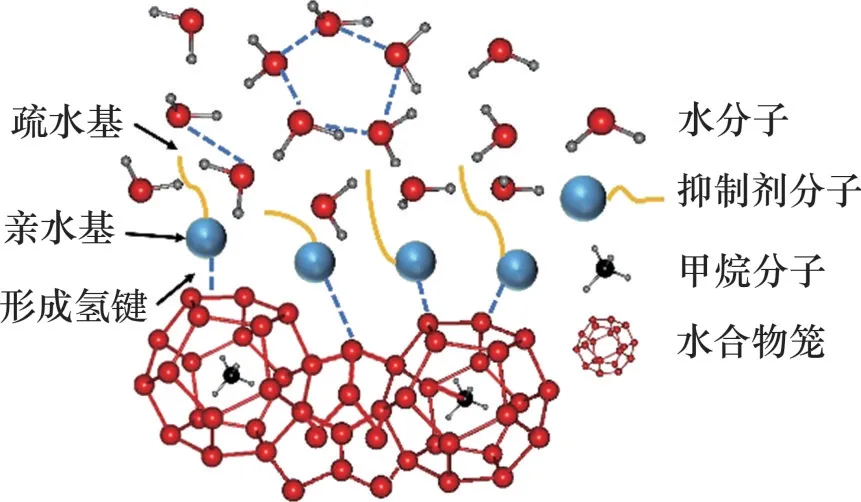
图1 吸附抑制机理示意图Fig.1 Schematic diagram of adsorption inhibition mechanism
1.3.2 扰乱抑制机理
水合物成核前即晶核达到临界尺寸时,抑制剂通过与周围的水分子形成氢键扰乱水分子有序结构,破坏了形成水合物笼形结构的关键团簇结构,并扰乱部分已形成的水合物团簇稳定结构。没有团簇结构,水合物无法成核,晶体也就难以形成。当水分子聚集成核后,抑制剂分子将吸附在水合物晶体的表面阻止水合物在某个方向上生长,如图2所示[21]。
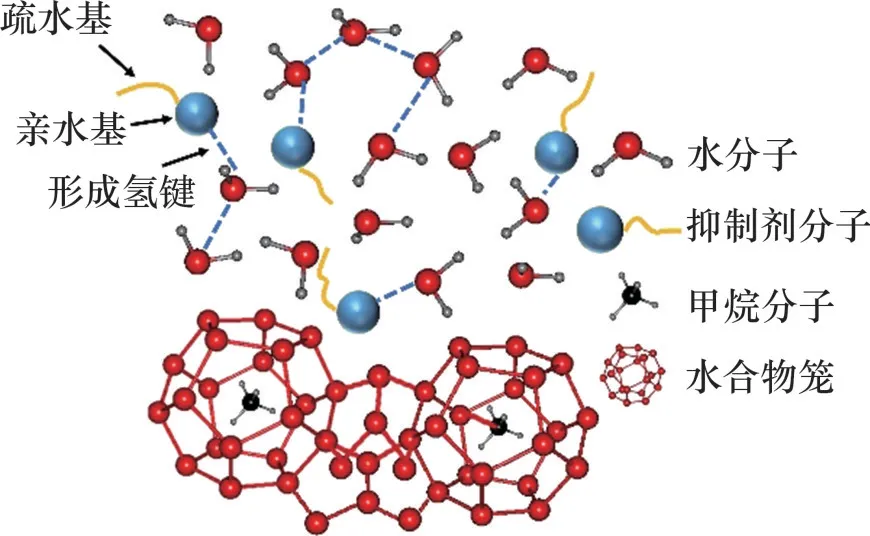
图2 扰乱抑制机理示意图Fig.2 Schematic diagram of disturbance suppression mechanism
1.3.3 层传质阻碍机理
当抑制剂质量分数足够大时,抑制剂分子会在气-液界面处形成聚合物层,阻碍水分子和客体分子的运动,增大分子的层传质阻力,从而抑制水合物的生长过程,如图3所示[22]。
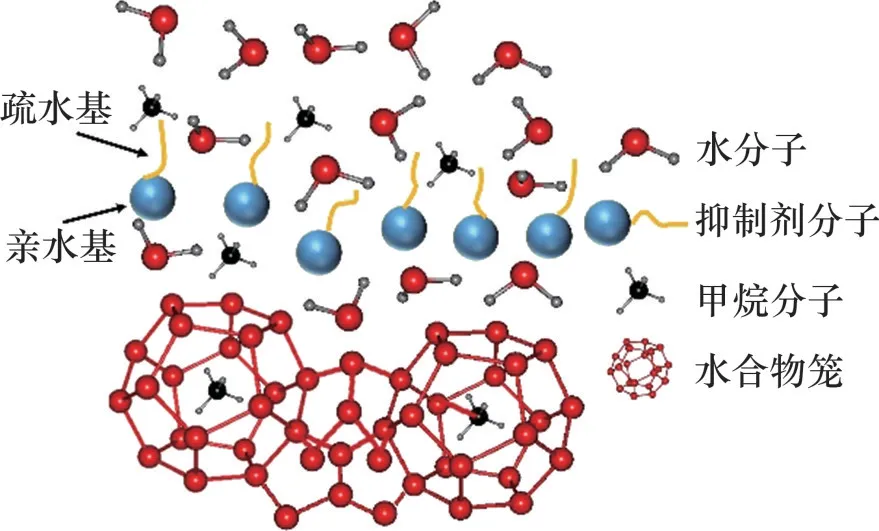
图3 层传质阻碍机理Fig.3 Mechanism of layer mass transfer hindrance
2 动力学抑制剂官能团抑制机理
自从筛选出以聚乙烯吡咯烷酮(PVP)为代表的第一代动力学抑制剂以来,研究者以第一代抑制剂为基础进行构效分析和设计改进,对动力学抑制剂分子结构特别是以功能性官能团为单位进行设计改进,并借助分子动力学模拟、分子设计等手段开发一系列具有较好抑制性能的动力学抑制剂,如图4所示。下面分别对这几类动力学抑制剂结构和性能进行总结与归纳。
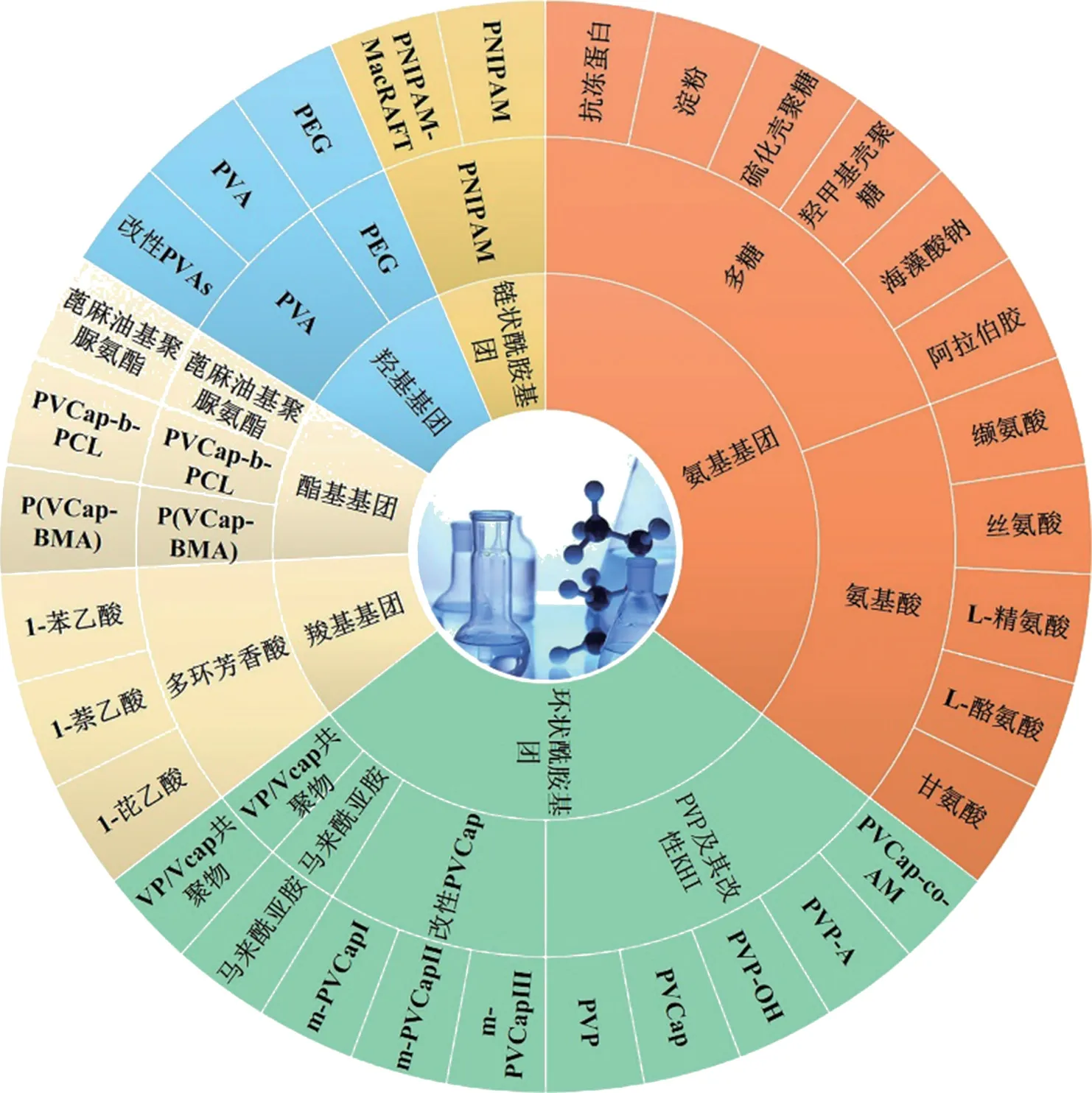
图4 各类官能团及相应抑制剂Fig.4 Various functional groups and corresponding inhibitors
2.1 环状酰胺基团
在水合物晶核成核前,水分子会自发形成“笼型团簇化”结构,环状酰胺类抑制剂分子上的酰胺基团与水分子相互作用形成氢键,扰乱水分子的有序结构,使得“笼型团簇化”难以长大成笼型。体系中不可避免地存在部分能够长大的“笼型团簇化”结构,当其尺寸长至临界尺寸时,环状酰胺类抑制剂分子上的酰胺基与该晶核的水分子作用,使其失去稳定性。如若体系中有少量晶核能够稳定存在,则抑制剂分子会吸附至生长面,且结合自由能越大的聚合物越容易吸附(抑制剂结合自由能为抑制剂体系总能量减去表面能量与抑制剂能量之和)[21],酰胺基上氧原子与水分子形成的氢键会破坏水合物笼原有的氢键和笼结构,聚合物在空间上形成的位阻也会阻碍水合物笼进一步生长[23],如图5所示。另外,当抑制剂溶解在水中后,环状酰胺基团的作用将促使抑制剂扩散并吸附在气-水表面,最终在气-水界面形成抑制剂分子层,阻碍主客体分子之间的传质过程,进一步延缓水合物生长[22]。
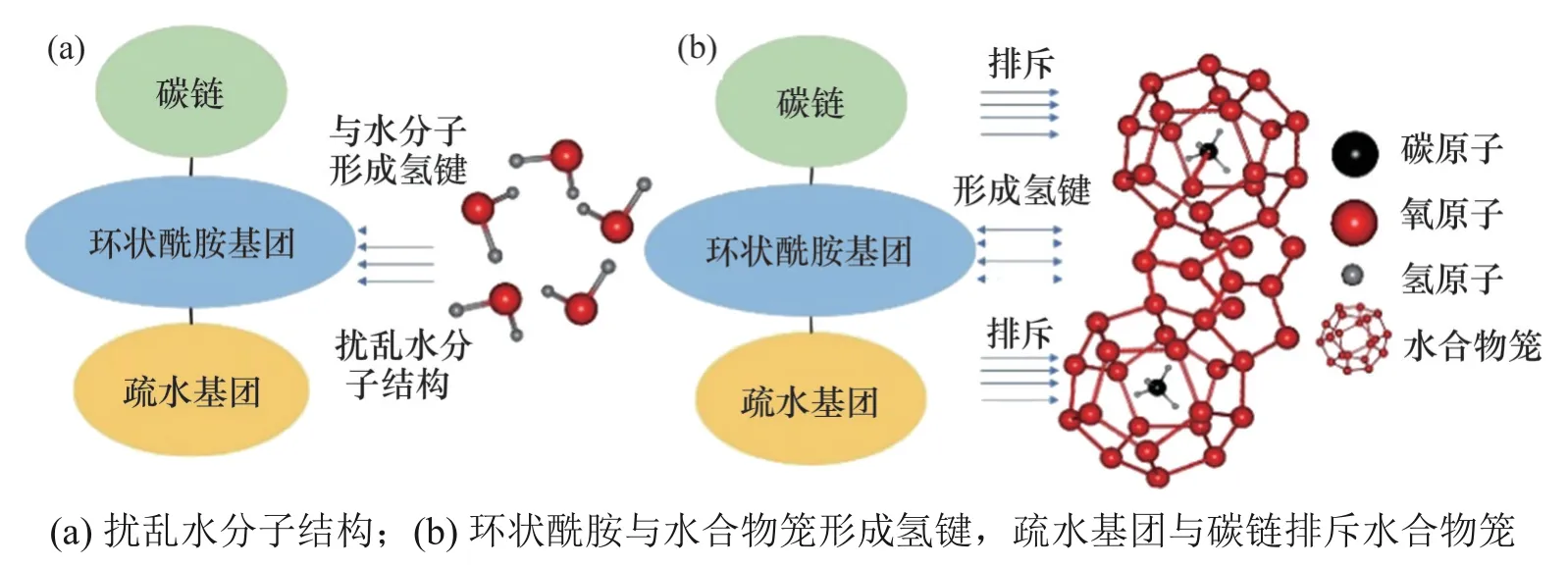
图5 环状酰胺基对水合物的抑制作用Fig.5 Inhibition of cyclic amide group on hydrate
2.1.1 聚乙烯基吡咯烷酮(PVP)
聚乙烯基吡咯烷酮(PVP)是最早筛选研发应用的环状酰胺类动力学抑制剂,其相对分子质量一般在10 000~3 5000 kDa之间[24],分子结构如图6所示。其衍生物都具有延长水合物诱导时间、降低水合物生长速率的抑制能力。研究表明,PVP主要通过环上酰胺基的双键氧与水分子形成氢键扰乱水分子有序结构,并且PVP 会吸附到水合物晶体表面抑制水合物的生长[25]。LI等[26]发现吡咯烷酮环的双键氧之所以能够与水分子形成氢键抑制水合物生长,是因为双键氧原子的高电荷密度和静电分子势(ESP)导致其对周围水分子具有强吸引力(如图7(a)所示);此外,由于吡咯烷酮环与甲烷(CH4)分子间的相互作用,导致大量CH4从H2O-CH4过饱和溶液中逸出,气泡的形成和水合物核附近CH4质量分数的降低减缓了CH4向水合物核表面的迁移和水合物的生长速度。CHENG 等[27]的研究表明,在水合物成核阶段,PVP能够干扰水分子间氢键并在空间上阻碍水合物笼的形成,但这并不是抑制水合物成核的主要原因,PVP主要通过加速甲烷与水分子的运动干扰水合物笼的稳定性,从而抑制水合物成核。LIU等[28]通过计算PVP,PVip和PVCap等抑制剂的KHIs·(H2O)n团簇分子的稳定能(Estab)对其稳定性进行评估,并将其与纯水团簇的稳定能进行比较,发现KHIs·(H2O)n的稳定能小于(H2O)n+1的稳定能。此外,计算水在KHIs·(H2O)n团簇中的扩散系数发现,与纯水团簇相比,抑制剂的加入使水分子更快地扩散,导致KHIs·(H2O)n团簇的稳定性降低,因此,推断PVP 是通过破坏水分子的局部组织以抑制水合物的形成。
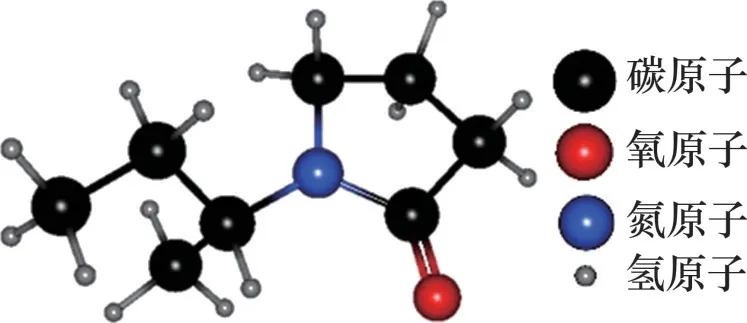
图6 PVP分子结构Fig.6 Molecular structure of PVP
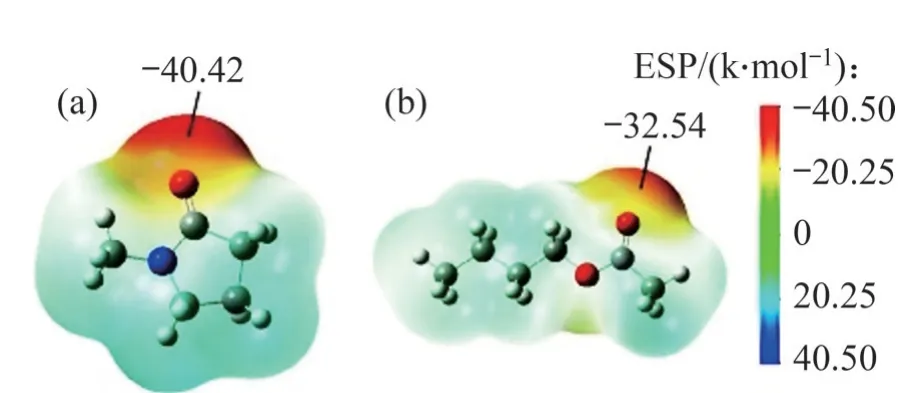
图7 不同抑制剂分子的静电势[26]Fig.7 Electrostatic potential of different inhibitor molecules[26]
环境温度对PVP的抑制产生显著影响,PVP过冷度约为5 ℃,在相对较高的温度下才能发挥抑制作用,当温度较低时,可能起促进作用[29]。此外,环上含碳数目也是影响PVP抑制性能的因素之一,在一定范围内,酰胺环上-CH2-数目越多,抑制能力越强[30]。环状酰胺类抑制剂的环尺寸同样影响PVP 抑制性能的发挥,计算水分子在KHIs·(H2O)n团簇中的扩散系数发现,随着环尺寸增大,溶液中水分子的扩散程度减弱,抑制剂的破坏作用会随着环尺寸增大而增强,更有效地抑制水合物笼的出现,并且环尺寸的增大可以提高聚合物与水合物笼的结合能力[28]。
2.1.2 聚乙烯基己内酰胺(PVCap)
由于环状酰胺类抑制剂性能与酰胺环上含碳数目在一定范围内呈正相关,因此,以PVP 分子结构为基础,增大环尺寸得到PVCap,分子结构如图8所示。LIU 等[28]的研究结果表明,PVCap 与PVP 内酰胺环相比尺寸增大,水分子五角环受到更大破坏,更有效地抑制了水合物笼的出现,如图9所示。不同抑制剂在开放式512笼上的平衡吸附构型[27]如图10所示(512是一种水合物笼结构,表示由12个五边形组成的12面体)。五角环中的水分子在PVP 系统模拟过程中可以保持稳定的氢键(见图10(a)),表明PVP 可能无法有效干扰五边形环,表现出较差的扰动抑制性能。而从图10(c)可知,在PVCap 作用下,水环更快地破裂,并且由于内酰胺环大小(即环上碳原子数目)不同,PVP只与笼状体的水分子形成1个氢键,而PVCap分别与笼状体上的水分子以及溶液中的水分子形成氢键,具有更强的抑制能力。此外,抑制剂和水合物之间的范德华力随着内酰胺环尺寸(即环上碳原子数目)的增加而增加,从而产生更好的抑制效果。
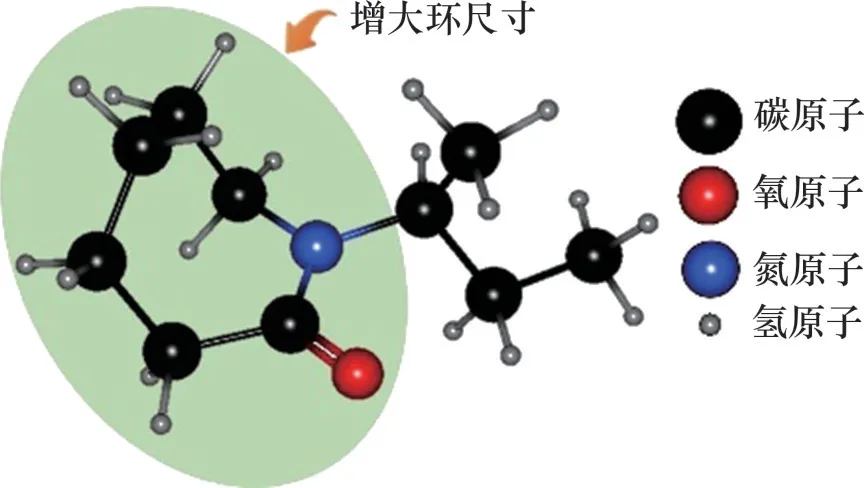
图8 PVCap分子结构Fig.8 Molecular structure of PVCap
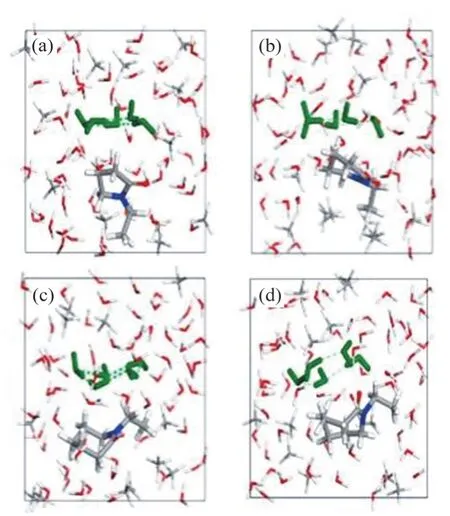
图9 五边形水环和不同抑制剂间相互作用的分子动力学模拟[28]Fig.9 Molecular dynamics simulation of interaction between pentagonal water ring and different inhibitors[28]
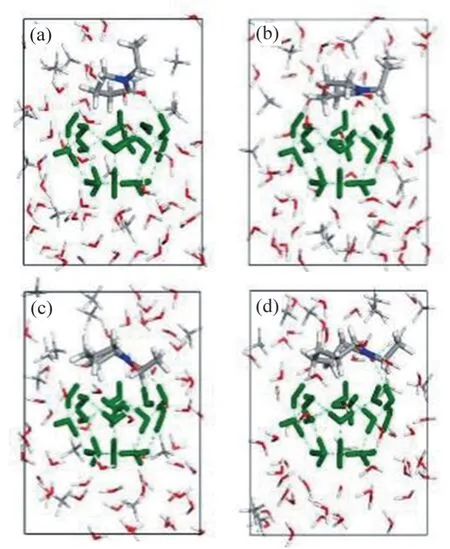
图10 不同抑制剂在开放式512笼上的平衡吸附构型[28]Fig.10 Equilibrium adsorption configuration of different inhibitors on he open 512 cage[28]
不同相对分子质量的PVCap 显示出不同抑制能力,低相对分子质量的PVCap 具有更强的干扰水分子生长的能力,而相对分子质量较高的PVCap 在长时间存在下能够与水合物表面产生更显著的吸附作用,通过覆盖未被占据的水合物孔穴具有更强的抑制水合物生长能力,但过高的相对分子质量的PVCap 会使聚合物分子链相互缠结无法覆盖水合物孔穴,导致抑制能力下降[31]。
2.1.3 端羟基聚己内酰胺(PVCap-OH)
端羟基聚己内酰胺(PVCap-OH)是以PVCap 抑制剂为基础,通过在其内酰胺环上引入羟基以提高抑制能力[32],其分子结构如图11所示。羟基基团的引入增强了亲水性,使其更容易与水分子形成氢键,破坏水分子的有序结构,PVCap-OH末端的羟基增强了聚合物与水分子的相互作用,阻止了气体分子的进入,导致诱导时间更长[33]。此外,较小的亲水性羟基基团可以插入到水分子结构中,在水合物成核阶段,阻止客体分子进入,从而延迟了水合物成核,降低了水合物生长速率[34]。另外,在PVCap 结构中引入羟基也显著增加了抑制剂-水合物的相互作用能,产生更好的抑制性能[32]。WAN等[33]发现羟基的引入增强了聚合物的亲水性。由于浊点性能取决于聚合物的亲水性[35-36],因此,羟基的引入使浊点降低,过冷度提高。除此之外,在PVCap 的聚乙烯链中引入羟基,增强了聚合物与水分子的交互性,醇羟基的引入大大提高了PVCap 的生物降解性,使得它更好地被环境所降解。
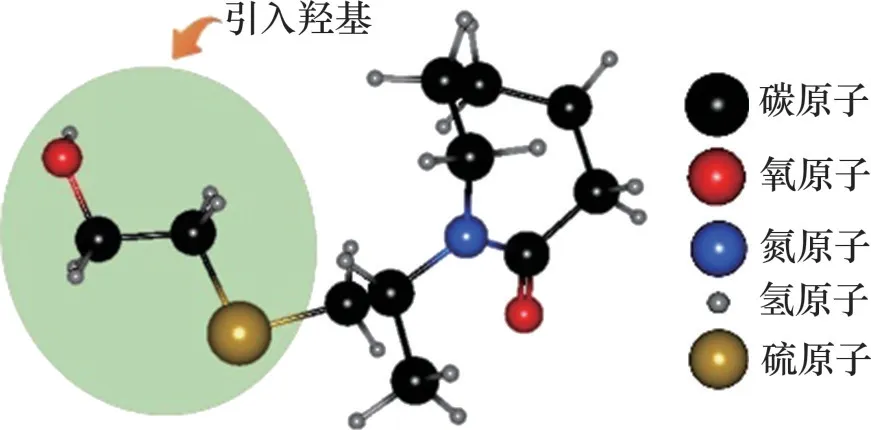
图11 端羟基聚己内酰胺(PVCap-OH)分子结构Fig.11 Molecular structure of hydroxyl-terminated polycaprolactam(PVCap-OH)
2.1.4 聚乙烯吡咯烷酮-α(PVP-A)
聚乙烯吡咯烷酮-α(PVP-A)是基于PVP 的一种酰胺类衍生物抑制剂,通过引入酯基与长链烷基提高PVP 的KHI 性能,其分子结构如图12所示。LI 等[37]发现PVP-A 通过干扰自由水分子在水合物表面的聚集来阻止水合物核达到自发生长的临界核尺寸,另外,它与水分子间的强结合效应极大地破坏了水分子与水合物表面之间氢键的作用,双键氧的强吸引力促使更多的水分子与抑制剂分子形成氢键,而不是让水合物继续生长,显著抑制了水合物核的生长,延长了水合物的生长时间。LI 等[26]还发现由于PVP-A 的酯基、吡咯烷酮环都能与周围的水分子形成较强的氢键,同时,长链烷基具有强疏水性,使得侧链烷基向气相延伸,导致PVP-A 分子的骨架在气液表面垂直排列,一部分浸入水中,另一部分在气相中伸展,如图13(a)所示。由于长链酯基和甲烷分子之间具有较强的相互作用,能够吸附H2O-CH4过饱和溶液中的甲烷分子,从而形成气泡,与此同时,伸展到甲烷气相中的侧链烷基与形成气泡的甲烷分子间具有强烈的相互作用,导致CH4分子很难再溶解到上层水溶液中,降低了水合物生长速率,抑制了水合物的生长。
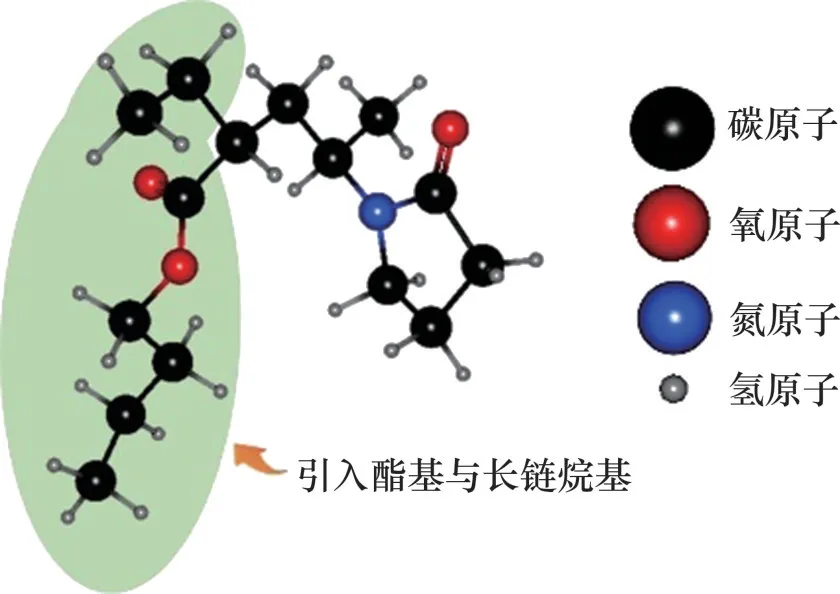
图12 PVP-A分子结构Fig.12 Molecular structure of PVP-A
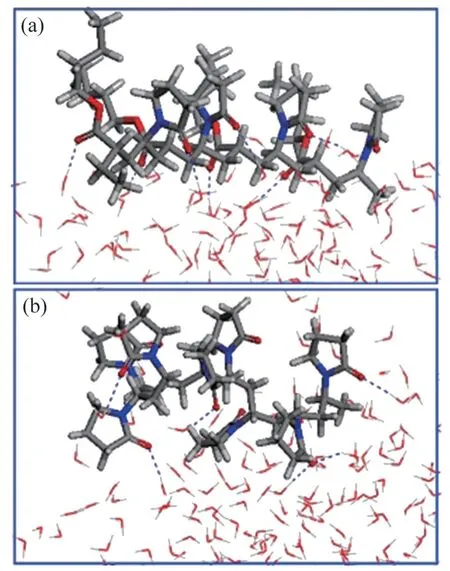
图13 含不同抑制剂系统的局部放大图[37]Fig.13 A partial enlarged view of system containing different inhibitors[37]
水溶液中动力学抑制剂聚合物的构象对其抑制性能起着至关重要的作用[26]。动力学抑制剂通常是同时具有亲水与疏水链段的两性聚合物,如图14所示。当抑制剂处于液相(动力学模拟阶段1)时,疏水基团倾向于聚集在一起,使亲水基团暴露于水相中,从而最大程度地降低整个分子的疏水性。对于PVP 分子,疏水性碳原子环的聚集使整个骨架发生不可逆弯曲,而对于PVP-A 分子,由于长链烷基具有强柔韧性,疏水性基团聚集在一起后并不会使整个分子的骨架发生变形。甲烷气泡形成后,抑制剂分子停留在气-液界面,亲水基团浸入液相,疏水基团延伸到气相。其中,PVP-A 分子骨架在气液界面得到充分伸展并有序排列,促使双键氧原子与水分子、疏水基团与甲烷分子之间充分接触,增强了抑制性能,而对于PVP 分子,不可逆的弯曲骨架削弱了整个PVP 分子的疏水性,减小了疏水基团与甲烷分子的接触面积,导致其不能最大限度地发挥抑制作用。此外,过冷度影响着PVP-A 抑制性能的发挥,当过冷度过高时,PVP-A 抑制能力丧失,甚至促进水合物的形成。

图14 PVP和PVP-A的分子构象以及不同阶段含KHIs体系的动力学模拟局部放大图[26]Fig.14 Molecular conformation of PVP and PVP-A and partial enlarged view of dynamic simulation of the KHIscontaining system at different stages[26]
2.1.5 聚(N-乙烯基己内酰胺-丙烯酰胺)(PVCap-co-AM)
LONG等[38]为获得更高效、更高溶解度的动力学抑制剂,在PVCap 基础上采用反向原子转移自由基聚合方法合成了一种新型嵌段共聚物抑制剂聚(N-乙烯基己内酰胺-丙烯酰胺)(PVCap-co-AM),其分子结构如图15所示。与水、PVP 及PVCap 相比,PVCap-co-AM 呈现较高的过冷温度,并且延迟水合物成核的时间更长。由于聚(N-乙烯基己内酰胺-丙烯酰胺)具有2 种不同的单体单元,因此,其链结构更加不规则,并且这2个单体单元使共聚物在溶液中的构象更加伸展,促使共聚物更多地与水分子相互作用。其次,在PVCap 共聚物中加入额外的亲水丙烯酰胺单体增加了共聚物与周围水分子相互作用的概率,增大了对水的扰动程度,因此,增强了对水分子构建水合物笼的阻碍作用,延缓了水合物成核时间。另外,冷冻扫描电镜图像显示PVCap-co-AM 会改变甲烷水合物的形态,形成的甲烷水合物不像致密堆积的饼状,而是像疏松的薄片,具有较大的孔隙率,随机分散在整个表面,如图16所示。
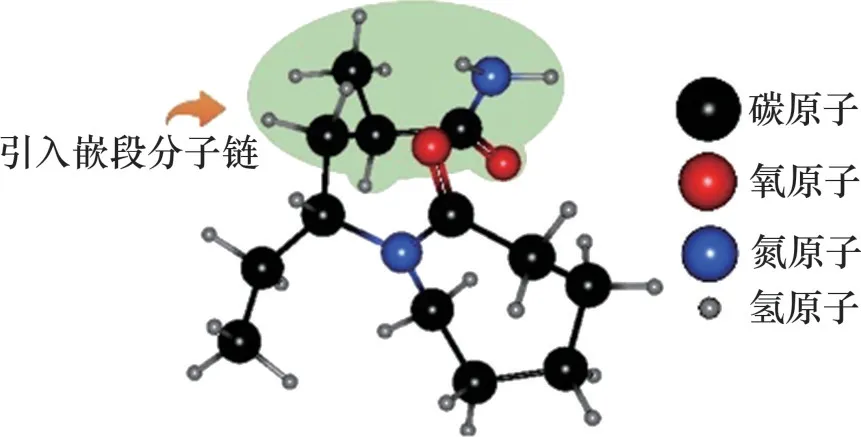
图15 聚(N-乙烯基己内酰胺-丙烯酰胺)(PVCap-co-AM)分子结构Fig.15 Molecular structure of poly(N-vinylcaprolactamacrylamide)(PVCap-co-AM)

图16 不同抑制剂存在下甲烷水合物表面微观结构的电镜扫描图像[38]Fig.16 Scanning electron microscope images of surface microstructure of methane hydrate in presence of different inhibitors[38]
较高质量分数的聚PVCap-co-AM 能显著延长诱导时间,PVCap-co-AM 质量分数与其抑制性能呈正相关。PVCap-co-AM 质量分数变化趋势与PVCap 和其他典型抑制剂质量分数的变化趋势相似,如图17所示。VP/VCap 共聚物分子结构见图18,改性PVA分子结构见图19。
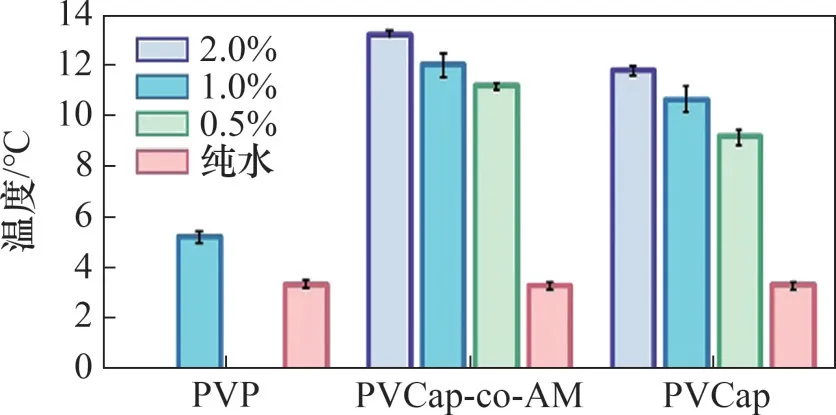
图17 不同质量分数的PVCap-co-AM过冷温度Fig.17 Supercooling temperature of PVCap-co-AM with different mass fractions
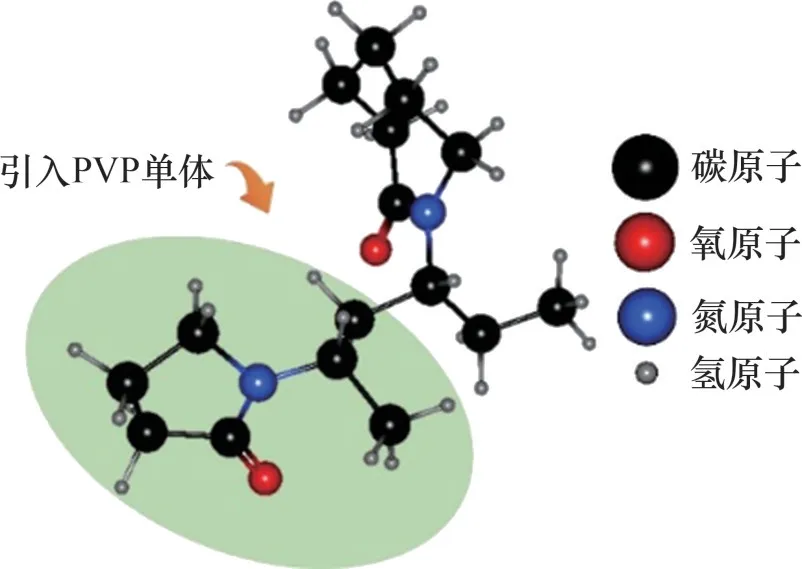
图18 VP/VCap共聚物分子结构Fig.18 Molecular structure of VP/VCap copolymer
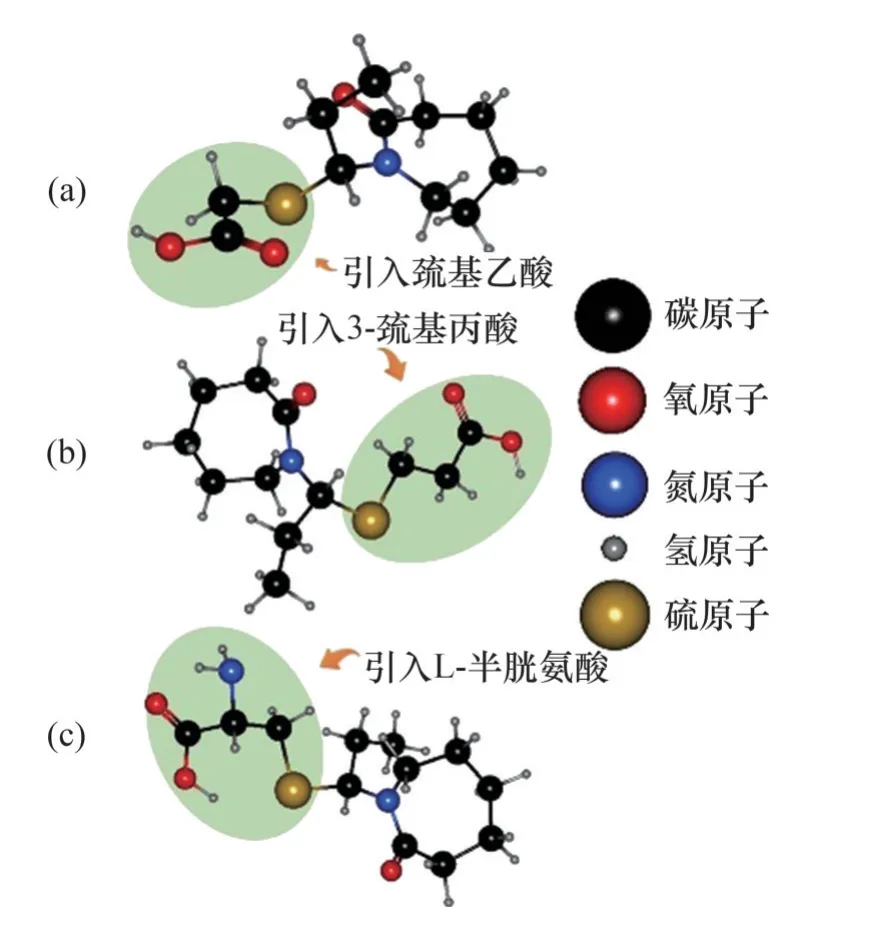
图19 改性PVA分子结构Fig.19 Molecular structure of modified PVA
2.2 VP/VCap共聚物
虽然PVCap 表现出比PVP 更好的KHI 性能,但PVCap的浊点Tcl通常为30~40 ℃[39],在高温水中的溶解度比PVP的低,因此,MOHSENZADE等[40]以PVCap 为基础引入亲水性PVP 单体进行聚合,得到了具有高浊点的有效动力学抑制剂,其分子结构如图20所示。而后基于获得的具有最佳性能VP/VCap 抑制剂,引入甲基丙烯酸二甲氨基乙酯(DMAEMA)合成三元共聚物。由于DMAEMA 官能团可以干扰水分子并抑制水合物的形成,因此,VP/VCap 的KHI 性能提高,过冷度与浊点提高。MOHSENZADE等[40]又引入甲基丙烯酸(MAA)和巯基丙酸(3-MPA)对VP/VCap改性,添加到改性共聚物链末端的3-MPA 官能团能够与水形成氢键,从而起到了更好的吸附和表面覆盖作用,提高了共聚物的KHI 性能。然而,MAA 的改性共聚物对水合物生长速率的影响较小,虽然MAA 的引入对KHI 性能没有改善,但浊点大大提高,而3-MPA恰恰相反,虽然显著增强了KHI 性能,但没有提高浊点。此外,3-MPA 的化学改性使得过冷度增加,MAA对最大过冷度的影响可以忽略不计。
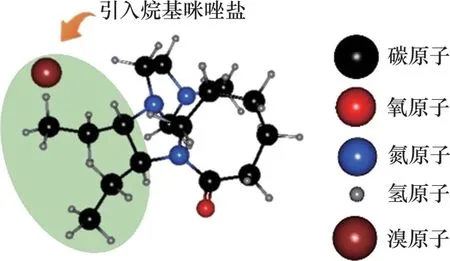
图20 1-乙烯基-3-甲基丁基咪唑溴化物共聚物分子结构Fig.20 Molecular structure of 1-vinyl-3-methylbutylimidazole bromide copolymer
VP 与VCap 抑制剂单体质量比对其抑制性能有显著影响。VCap 质量增加会提高其KHI 性能,当VP与VCap的质量比为1:3时,VP/VCap抑制能力比PVCap 的强;VP/VCap 的最大过冷度与单体质量相关。由于VP单体的亲水性,VP与VCap质量比增加能够增强VP/VCap的浊点。
2.3 改性PVCap
MOHSENZADE等[31]为获得合成路线简单、有效且浊点较高的KHIs,引入巯基乙酸、3-巯基丙酸、L-半胱氨酸3 种氨基酸合成了m-PVCap I,m-PVCap Ⅱ和m-PVCap Ⅲ,它们的分子结构如图19所示。从图19可见:这3种氨基酸化学结构中的羟基、氨基与氨基增强了改性PVCap的亲水性,从而获得了更高的浊点;此外,所有改性PVCap对SI和SⅡ水合物的抑制能力均比PVCap对SI和SⅡ水合物的抑制能力强,其中,m-PVCap I与m-PVCap Ⅱ分别对SI和SⅡ水合物显示出最佳的抑制性能。这是由于改性PVCap 不但可以像PVCap 一样通过羰基吸附到水合物表面,还可以通过引入的羟基结构与水合物表面的水分子形成更强的氢键。
2.4 1-乙烯基-3-甲基丁基咪唑溴化物共聚物
为了获得比PVCap 更高溶解度的抑制剂,以PVCap 为基础引入烷基咪唑盐进行聚合获得了一系列新型抑制剂[41],见图20。其中,聚[VCapV32 MBIMBr]能够阻止水合物的形成并且其拟制能力比PVCap 的抑制能力强,而聚[VCapV3BIMBr]、聚[VCapV3EIMBr]和聚[VCapV33MBIMBr]不能完全阻止水合物的形成,但阻止了水合物在系统内的聚集,如图21所示。此外,合成的聚合物中只有聚[VCapV33MBIMBr]在一定质量分数下才能使THF 水合物晶体生长。引入1-乙烯基-3-(2-甲基丁基)咪唑鎓、1-乙烯基-3-乙基咪唑鎓、1-乙烯基-3-丁基咪唑鎓与1-乙烯基-3-(3-甲基丁基)咪唑鎓基团后,聚[VCapV32MBIMBr]、聚[VCapV3EIMBr]、聚[VCapV3BIMBr]和聚[VCapV33MBIMBr]的亲水性与浊点相比于PVCap 得到明显提高,因而,其能够在苛刻的温度和盐度下作为高效的动力学抑制剂使用。
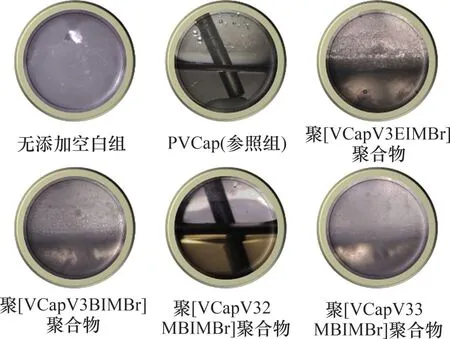
图21 各类聚合物存在下水-天然气系统的物理外观[41]Fig.21 Physical appearance of water-natural gas system in presence of various polymers[41]
2.5 链状酰胺基团
链状酰胺类抑制剂是一类在链结构中具有—N—C=O 基团的动力学抑制剂。酰胺含亲水性较强的—N=O 基团,—N=O 中的氮及氧原子电负性大,可以在水合物形成过程中与水合物笼上的氢原子形成氢键[42],其通常也有一些疏水末端,疏水部分(长碳链中的烷基基团和悬浮物基团)将排斥不稳定团簇水分子,抑制水合物的成核与生长,如图22所示。已证明许多酰胺基聚合物对气体水合物具有动力学抑制作用[43],这些聚合物主要包括丙烯酰胺聚合物[44]、乙烯基酰胺聚合物[45]和马来酰亚胺聚合物[46]。
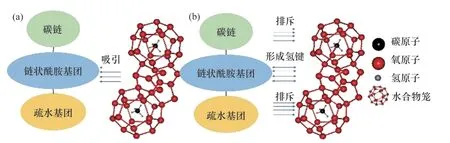
图22 链状酰胺基团对水合物的抑制作用Fig.22 Inhibition of chain amide group on hydrate
2.5.1 聚(N-异丙基丙烯酰胺)
聚(N-异丙基丙烯酰胺)(PNIPAM)是一种具有代表性的酰胺基聚合物,由于酰胺官能团连接在疏水基团上,因此显示出很强的动力学抑制(KHI)性能[47-48],其分子结构如图23所示。PARK 等[49]通过对水合物开始时间和过冷温度进行定量分析,发现合成的PNIPAM提高了水合物成核的过冷温度并延长了诱导时间。DA 等[35]合成了基于PNIPAM改性的动力学水合物抑制剂库,改性聚合物也显著拟制水合物生长,表明链状酰胺基与水合物晶体表面具有较强的相互作用。SEO等[50]也通过实验证实了PNIPAM抑制水合物的能力。
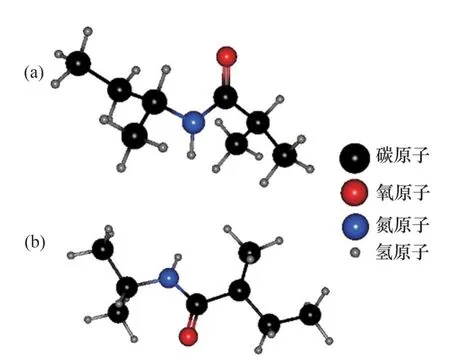
图23 聚合物分子结构Fig.23 Molecular structure of polymer
分子结构组成影响着抑制剂的抑制性能,PARK 等[49]发现侧基上烷基链长度影响PNIPAM 的抑制性能。无论是在自由水中还是在水合物颗粒表面,侧链长度增加都会使聚合物与水分子产生更大的范德华力,使抑制性能得到提高。此外,聚合物的立构规整度影响PNIPAM的抑制水合物性能,如图24所示。从图24可见有3 种类型的立构规整度,分别为所有侧基都位于主链一侧的全同立构、具有交替取向侧基的间同立构和具有随机取向侧基的无规立构。间规度为70%的PNIPAM比间规度较低的PNIPAM具有更好的抑制性能。基于成核抑制机制推测,间同立构结构所占比例增加增大了聚合物的链段长度,从而增强了对水的扰动,因此,对破坏水合物成核显示出更好的性能。另外,可能由于酰胺基团得到充分伸展,从而有更多的空间与水相互作用,间同立构所占百分比增加提高了PNIPAM(对于无规立构PNIPAM)的浊点。此外,PNIPAM与过苯甲酸叔丁酯(TBPB)存在协同抑制效应,PNIPAM与TBPB混合使用能够显著延长诱导时间,降低水合物生长速率[50]。
2.5.2 聚(N-异丙基丙烯酰胺)(PNIPAM-MacRaft)
PARK 等[5]使用可逆加成断裂链转移聚合(RAFT)合成了直链-PNIPAM 和支链-PNIPAM,通过与传统自由基聚合合成的PNIPAM 和市售KHIs(PVP 和Luvicap)进行比较以探究聚合物结构对抑制剂性能的影响,结果表明聚合物的结构是影响水合物抑制性能的重要因素之一。虽然线性PNIPAM显示出延迟水合物成核的最佳性能,但其不能有效抑制水合物晶体的生长。图25所示为水合物形成过程中捕获的图像,与线性类似物相比,支化聚合物结构能更好地与生长中的水合物晶体相互作用。扭矩可以用来估计存在水合物颗粒的烃流体的流动性[49],通过比较扭矩变化发现支化聚合物可以抑制水合物颗粒的团聚,减缓水合物的生长速度,因此,通过控制KHIs 的结构可以调节其抑制性能[35,49]。

图25 不同抑制剂存在下水合物形成过程中水合物相的图像[5]Fig.25 Images of hydrate phase in the hydrate formation process in presence of different inhibitors[5]
2.5.3 马来酰亚胺
由于马来酰亚胺原料来源广泛、合成简单、成本低,并且具有较强的抑制水合物能力,因此,是良好的动力学抑制剂。ZHANG 等[51]使用各类胺与马来酸酐均聚物合成了具有不同烷基的马来酰亚胺聚合物,以改进其性能,其分子结构如图26所示。结果表明,当马来酰亚胺侧链烷基中含有4~6个碳原子特别是含有5或6元环时,马来酰亚胺具有良好的抑制性能。此外,引入一定比例的二元胺(分别为3-二甲氨基丙胺(DMAPA)、N,N-二丁基乙二胺(DMEDA)与3-(二丁氨基)-1-丙胺(DBAPA)),使马来酰亚胺聚合物在水中的溶解能力与KHI性能得到提高,其中,DBAPA 的引入效果最显著。实验结果显示,在一定质量分数范围内,马来酰亚胺抑制水合物生长的能力随着其质量分数增加而增强。
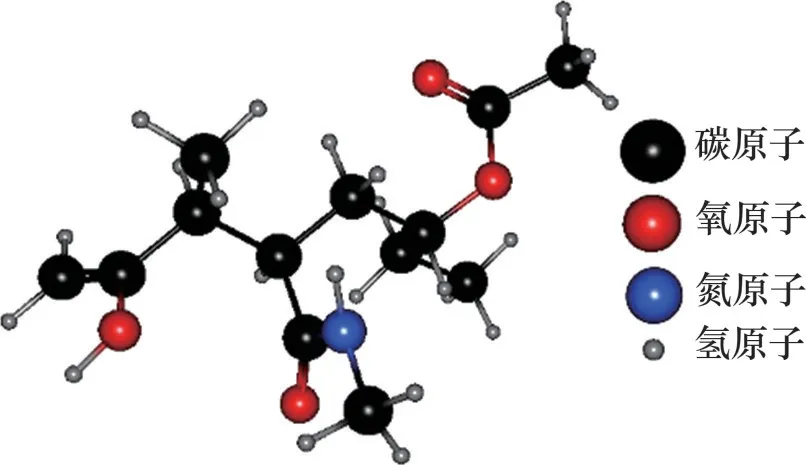
图26 马来酰亚胺分子结构Fig.26 Molecular structure of maleimide
2.6 羟基官能团
由于羟基是良好的氢键受体,因此,能与水分子形成氢键并固定水分子,扰乱周围水分子的结构;另外,羟基基团这样的小基团会占据客体分子在水笼内的位置,阻止气体分子进入,这些作用延迟了水合物成核并降低水合物生长速率羟基基团对水合物的抑制作用,见图27。
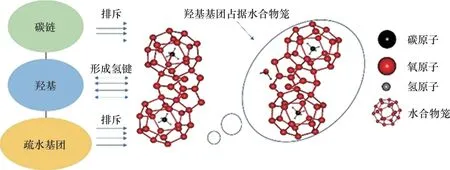
图27 羟基基团对水合物的抑制作用Fig.27 Inhibition of hydroxyl group on hydrate
2.6.1 聚乙二醇(PEG)
聚乙二醇(PEG)通常是亲水性强的非离子表面活性剂,其亲水亲油平衡值约为19,分子结构如图28所示。PEG 这类表面活性剂亲水部分会破坏其周围水分子之间的氢键,并且当相对分子质量较小时能够进入水合物孔穴,抑制水合物成核与生长[52]。虽然聚乙二醇能够起到抑制作用,但聚乙二醇性能不如商用抑制剂(PVP和Luvicap EG)的性能,这可能与抑制剂的结构和官能团有关。PVP这类聚合物的酰胺基团使其可以吸附在水合物表面,而聚乙二醇不含酰胺基团,不具有类似机制。
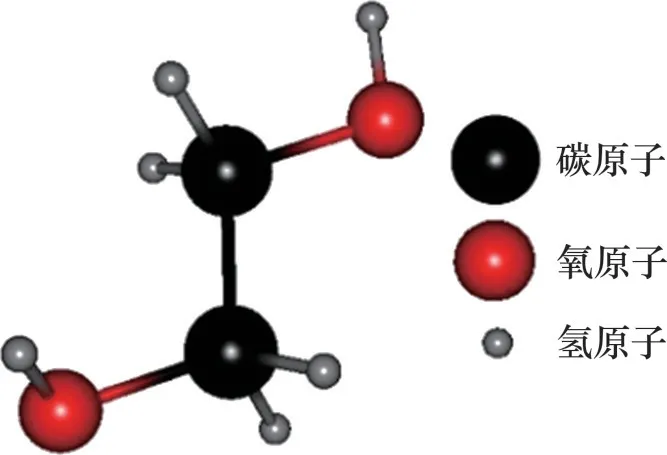
图28 聚乙二醇分子结构Fig.28 Molecular structure of polyethylene glycol
FOROUTAN 等[53]研究了不同相对分子质量的聚乙二醇的KHI 性能,结果表明相对分子质量较高的聚乙二醇(20 000)显示较弱的抑制作用,而低相对分子质量聚乙二醇(300)不但不具有抑制作用,反而提高了水合物的生长速度。经分析认为,相对分子质量较低的聚乙二醇(300)链长不足以覆盖水合物表面,无法抑制气体水合物形成,而相对分子质量较高的聚乙二醇(20 000)则可能具有这种能力并发挥抑制作用[54]。但总体来说,无论PEG相对分子质量高低,其对水合物生长速率的影响非常弱。此外,聚乙二醇质量分数、相对分子质量对其延迟水合物成核具有综合影响。聚乙二醇(相对分子质量为20 000)在质量分数为0.01%和0.10%时均能够延迟甲烷水合物形成的诱导时间,而聚乙二醇(相对分子质量为300)在质量分数为0.10%时加快了水合物成核速度。
聚乙二醇与PVP 存在一定的协同抑制作用,并且聚乙二醇质量分数和相对分子质量对聚乙二醇的协同抑制性能产生一定影响:质量分数为0.01%的聚乙二醇(相对分子质量为20 000)溶液可将PVP 的拟制能力提高27%~47%,但在聚乙二醇较高质量分数下,协同效应降低;质量分数为0.01%的聚乙二醇(相对分子质量为300)不但无法提高PVP 的性能,反而导致生长率增加,含质量分数为0.10%的聚乙二醇(相对分子质量为300)能够提高PVP的抑制性能,但效果很小[53]。
2.6.2 聚乙烯醇(PVA)
聚乙烯醇(PVA)是一种水溶性高分子聚合物,其分子侧链上含有大量羟基基团,PVA 在低质量分数下对晶体的成核有抑制作用,同时对晶体生长也有抑制作用[55],其分子结构如图29所示。TAKAAKI 等[56]通过实验发现聚乙烯醇具有抑制冰重结晶的能力。INADA 等[57-58]通过实验研究了PVA 对冰晶颗粒生长的抑制作用,发现PVA 在低质量分数下能够完全抑制冰晶的生长,认为由于PVA 支链羟基与水分子相互作用,PVA 极易吸附在晶体表面,从而抑制晶体进一步生长,而水合物是一种类冰的包合物,与冰的结构相似,因此,推测PVA 对水合物生长有一定抑制作用。实验证明聚乙烯醇对水合物成核具有较好抑制作用[59]。另外,JOKANDAN 等[60]发现PVA 与PVP 抑制剂具有一定协同抑制作用。
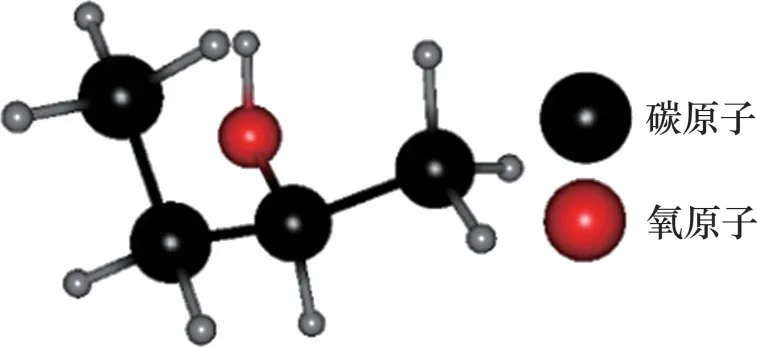
图29 聚乙烯醇分子结构Fig.29 Molecular structure of polyvinyl alcohol
PVA 质量分数是影响其抑制性能的重要因素之一[42],其抑制性能与质量分数不呈线性关系,而是存在一个最佳抑制性能的临界值。此外,PVA的相对分子质量、水解度也影响PVA 抑制性能,并且抑制性能与PVA 的相对分子质量、水解度存在线性增大的关系[56]。
2.6.3 改性聚乙烯醇(PVAs)
ROOSTA等[61]使用丙烯酰胺、甲基丙烯酰胺和丙烯腈对聚乙烯醇进行化学改性合成改性聚乙烯醇(PVAs),分别是将以上3种单体接枝到聚乙烯醇上得到接枝共聚改性PVAs 即PVA-g-AM,PVA-g-MAM 和PVA-g-AN。使用3 种单体官能化聚乙烯醇得到的官能化改性PVAs 为PVA-AM,PVAMAM 和PVA-AN。其中,接枝共聚改性虽然提高了PVA的抑制性能,但与PVP相比性能并不突出,而官能化得到的改性聚乙烯醇具有良好的抑制性能。在改性PVAs 中,PVA-AM 抑制性能最好,并且相比于PVP 抑制剂,PVA-AM 具有更好的抑制性能。其他官能化PVAs(PVA-MAM 和PVA-AN)也具有良好的抑制性能,如图30所示。研究表明,改性PVAs上的酰胺和腈官能团可以与水合物晶体表面的水分子形成大量较强的氢键,使聚合物在水合物表面产生更好的吸附作用,吸附的分子堵塞了水合物的未完成的空穴(作为活性生长位点),使客体气体分子(如甲烷和丙烷)难以进入这些空穴,从而抑制水合物生长。另外,表面张力降低不利于抑制水合物的生长(表面张力降低导致气-液界面的传质阻力降低,随后液相中的气体吸收速率增加,导致水合物形成速率增加[62]),而PVA 的表面张力小于比纯水的表面张力小,用甲基丙烯酰胺和丙烯腈对聚乙烯醇改性后增加了表面张力,从而提高了抑制性能。
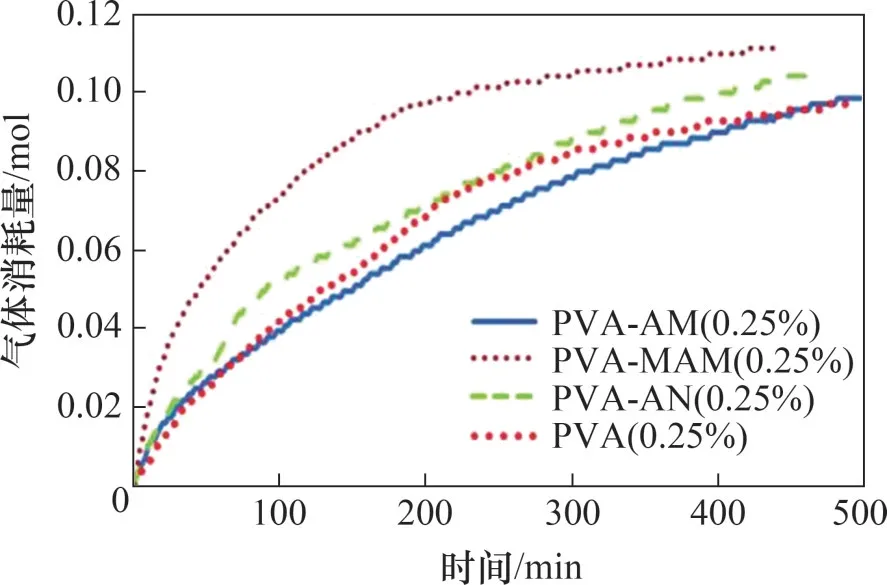
图30 官能化PVAs和PVA对水合物生长抑制作用的比较Fig.30 Comparison of inhibitory effects of functionalized PVAs and PVA on hydrate growth
改性聚乙烯醇相对分子质量影响其抑制性能。丙烯酰胺在主链聚合物上的接枝共聚会产生具有相对分子质量非常高的聚合物,而聚合物相对分子质量增大会降低动力学抑制剂的抑制作用[63],导致接枝共聚改性PVAs 与官能化改性PVAs 在性能上出现差异。另外,改性PVAs的性能也取决于其分子结构,PVA-AM和PVA-MAM具有非常相似的分子结构,区别只在于PVA-MAM具有额外的—CH3基团。然而,额外存在的—CH3基团降低了水-水合物界面亲水基团的密度,改变了界面处水分子质量分数,使PVA-AM(作为强抑制剂)和PVA-MAM(作为弱抑制剂)性能产生明显差异。与此同时,随着改性PVAs聚合物质量分数增加,其在水合物表面产生了的吸附作用更强,导致其抑制性能增强。
2.7 氨基官能团
位于水合物笼上的水分子中的氧原子带负电,相应的氢原子带正电,氨基中的氮原子带正电,而相应的氢原子带负电,因此,氨基中的氮原子能够与水分子中的氢原子结合,氢原子能够与水分子中的氧原子结合。氨基与水合物笼的稳定吸附方式有2种:一是氨基中的氢原子与水合物表面的氧原子相连;二是氨基中的氢、氮原子同时吸附在水合物表面的氧原子和氢原子上[64]。氨基通过这2种方式吸附在水合物表面上,达到抑制水合物的目的。
2.7.1 氨基酸
氨基酸是构成地球生命的重要组成部分,包括20 种不同的天然分子,每种分子都具有1 个羧酸、1 个氨基和1 个独特的侧链基团,它们相结合构成了生物体中的蛋白质。氨基酸具有环境友好、生物降解能力强的特性[65]。虽然现有的内酰胺单体的聚合物类动力学抑制剂显示出较强的抑制能力,但它们的生物降解能力低且抑制活性较弱[66]。与此同时,氨基酸具有以下特殊性质:在水合物形成过程中,大多数氨基酸表现为两性离子(即在溶液中既能与氢离子又能与氢氧根离子反应的离子)[67],使得它们能够与水分子发生强静电作用,电荷周围的水分子的有序结构受到破坏[68-69]。尽管烷基侧链的存在使氨基酸具有一定疏水性,但由于也具有羧酸和氨基基团,氨基酸也同时表现出亲水性,因此,氨基酸可以通过氢键与水分子相互作用。而最近的研究表明,动力学抑制剂对水合物的作用是由亲水与疏水性引起的[70]。BHATTACHARJEE等[71]发现氨基酸与水分子间的相互作用越强,抑制性能越好。由于氨基酸的亲水性和疏水性之间的平衡容易控制,因此,具有经济性、环保性、可回收性的氨基酸被开发成为新型动力学抑制剂。MADDAH 等[72]发现氨基酸和水合物晶体表面之间的范德华力作用和静电作用抑制了水合物的生长,并且氨基酸的加入减少了甲烷在水中的扩散。MADDAH 等[72]还发现氨基酸侧链长度影响氨基酸的抑制性能,存在决定动力学抑制性能的临界烷基链长度,并且在许多实验中都观察到这一点。此外,氨基酸还通过物理吸附的方式与水合物表面相结合以达到抑制水合物的目的,即氨基酸亲水基团中的氧原子、氮原子与水分子中的氢原子结合,氨基酸亲水基团中的氢原子与水分子中的氧原子结合[64]。此外,氨基酸的质量分数影响氨基酸的抑制能力,氨基酸质量分数越高对甲烷水合物生长的抑制效果越好。氨基基团对水合物的抑制作用如图31所示。
2.7.2 丝氨酸、缬氨酸
HU 等[73]进行分子动力学模拟时发现丝氨酸、缬氨酸通过与水分子形成氢键,扰动水分子结构,影响水合物笼的形成,从而延迟甲烷水合物的成核和生长。此外,由于丝氨酸具有额外的羟基基团,使其具有较高的溶解度,因此,可以与水分子形成更多氢键,显示出更强的抑制性能[72],丝氨酸和缬氨酸分子结构如图32所示。此外,他们还发现丝氨酸、缬氨酸倾向通过3种方式吸附于水合物笼上,分别为:通过亲水的羟基基团中的氢原子、氧原子同时与水合物笼的氧原子、氢原子相结合;通过亲水的羧基基团中的氢原子、单键氧原子与水合物笼的氧原子氢原子结合;通过亲水的羧基基团中的氢原子、双键氧原子与水合物笼的氧原子、氢原子结合。其中,缬氨酸的羧基作用最强,但由于缬氨酸的疏水性甲基侧链破坏了亲水部分周围水分子间的氢键,使得亲水基团与水分子间作用减弱,抑制性能下降,其拟制能力比丝氨酸的弱[64]。
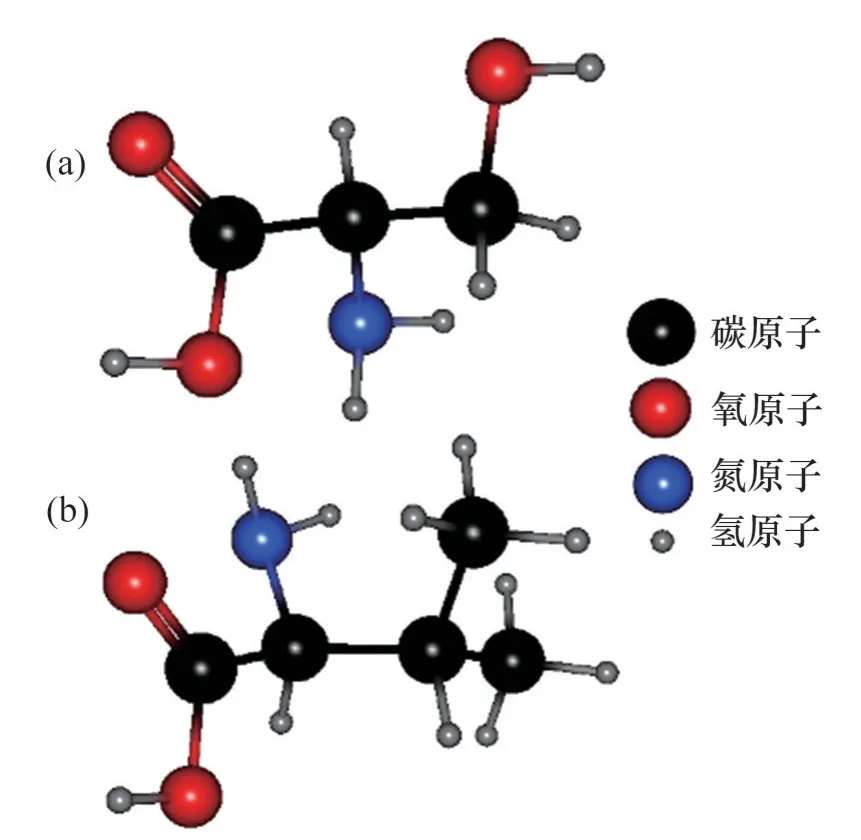
图32 氨基酸分子结构Fig.32 Amino acid molecular structure
氨基酸在水中的溶解度是影响其抑制性能的重要因素,丝氨酸由于疏水性低,在水中溶解度高,具有很多用于形成氢键的供体和受体原子,并且由于没有环状结构,因此,丝氨酸分子能够充分伸展,具有较强的抑制性能。此外,丝氨酸、缬氨酸的抑制能力随着它们质量分数增大而增强。
2.7.3 L-酪氨酸
L-酪氨酸能够抑制水合物的成核,延长水合物形成的诱导时间,并且对水合物成核后的进一步生长也具有抑制作用[74],其分子结构如图33所示。L-酪氨酸通过干扰局部水结构增加成核障碍,或者通过氢键吸附在水合物晶体上,达到抑制水合物成核与生长的目的。
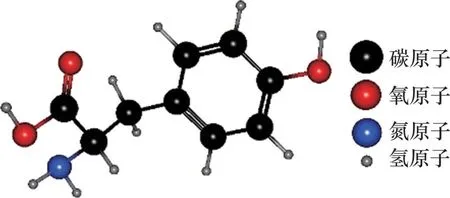
图33 L-酪氨酸分子结构Fig.33 Molecular structure of L-tyrosine
在水合物形成过程中,L-酪氨酸作为抑制剂与PVP 抑制剂存在一定协同抑制作用,酪氨酸的存在能够增强PVP 的抑制性能,如图34和图35所示。
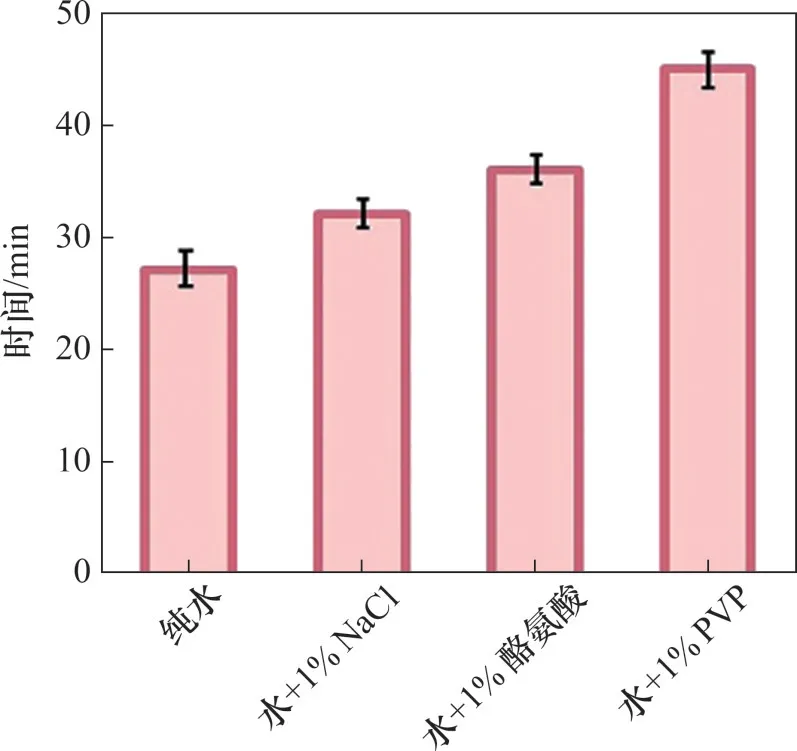
图34 氯化钠、左旋酪氨酸和聚乙烯吡咯烷酮存在时水合物形成的诱导时间Fig.34 Induction time of hydrate formation in presence of sodium chloride,L-tyrosine and polyvinylpyrrolidone
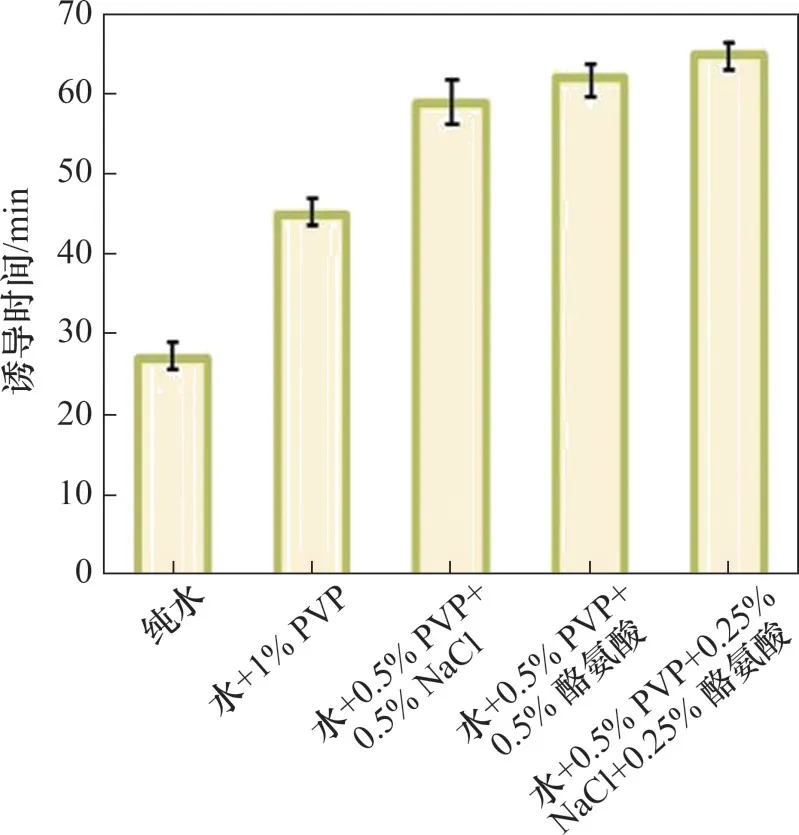
图35 氯化钠、左旋酪氨酸作为抑制剂与聚乙烯吡咯烷酮产生协同作用时水合物形成的诱导时间Fig.35 Induction time of hydrate formation when sodium chloride and L-tyrosine as inhibitors have synergistic effect with polyvinylpyrrolidone
2.7.4 L-精氨酸、甘氨酸
WANG 等[75]通过实验发现,氨基酸的亲水性会对水分子产生干扰,达到抑制水合物成核与生长的目的[76],且氨基酸亲水性影响其对水合物成核与生长的抑制能力[77]。然而,L-精氨酸亲水性比甘氨酸的强,但由于甘氨酸的分子链比L-精氨酸短,其分子结构更接近水分子结构,分子自身之间的摩擦和相互作用很弱,使其对水分子的干扰比对L-精氨酸的干扰更强,即在相同的浓度下,当2个亲水性氨基酸的分子数相同时,结合水分子能力较弱的甘氨酸可能会干扰水分子的有序结构,使水分子更难产生“水笼”包裹客体分子形成水合物,因此,甘氨酸的水合物抑制能力更强;而当浓度相同时,甘氨酸溶液的分子数可能是左旋精氨酸溶液的2.3 倍,甘氨酸均匀分布在这些溶液中,可以对水分子产生更有效的干扰。此外,甘氨酸具有消除水合物记忆效应的能力(记忆效应是一种现象,在这种现象中,曾形成过水合物的气体和水更容易再次形成水合物)[78]。两者分子结构如图36所示。
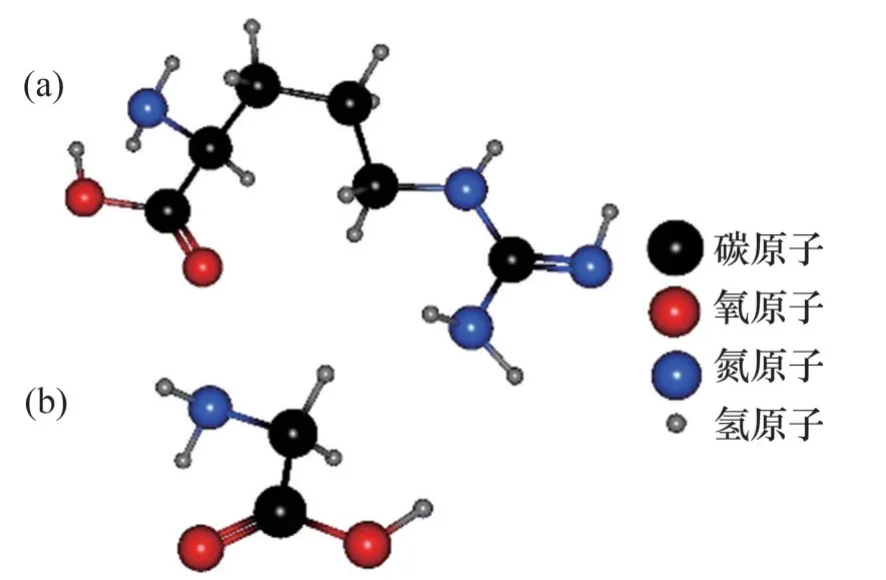
图36 氨基酸分子结构Fig.36 Molecular structure of amino acid
不同质量分数的甘氨酸和L-精氨酸对水合物的形成表现出不同程度的抑制能力,通过测量2种不同质量分数的氨基酸水溶液发现溶液的导热系数随着氨基酸体积的增加而降低,抑制能力随之增强。另外,甘氨酸和L-精氨酸的剂量影响其抑制性能。对于甘氨酸,其使用剂量存在1 个临界值,当甘氨酸使用剂量小于该临界值时,甘氨酸对水分子的干扰可能主导其对水合物形成的影响,水分子很难形成笼状结构,从而抑制水合物的形成;然而,当甘氨酸使用剂量超过该临界值时,在系统中受到甘氨酸干扰的水分子变得越来越活跃。BHATTACHARJEE 等[79]研究表明,随着甘氨酸剂量增加,它可能不影响或促进水合物的形成。而对于L-精氨酸,随着其用量增加,其抑制性能逐渐增强,L-精氨酸通过与水分子结合抑制水合物生长的作用一直占主导地位。各组试验水合物形成的诱导时间和过冷度见图37。
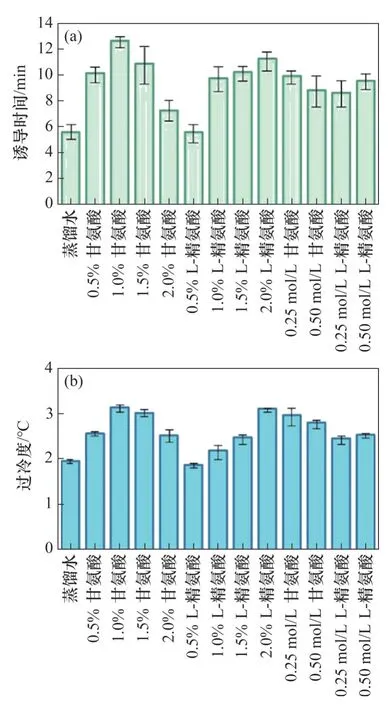
图37 各组试验水合物形成的诱导时间和过冷度Fig.37 Induction time and supercooling degree of hydrate formation in each group of test
WANG 等[75]还发现,甘氨酸与L-精氨酸与PVPK90 具有良好的协同抑制作用,如图38所示。当使用甘氨酸或L-精氨酸与PVPK90的组合时,由于其具有亲水性,因此,可以通过干扰和结合水分子来阻止水分子的定向排列形成笼状结构,而当部分水分子在氢键作用下形成半笼状结构或笼状结构即“水笼”的体积足够大时,PVPK90将吸附在其表面,阻止THF 分子进入这些笼状结构,或阻止水分子、THF 分子和孤立水合物晶粒之间进一步接触[80-81],如图39所示。
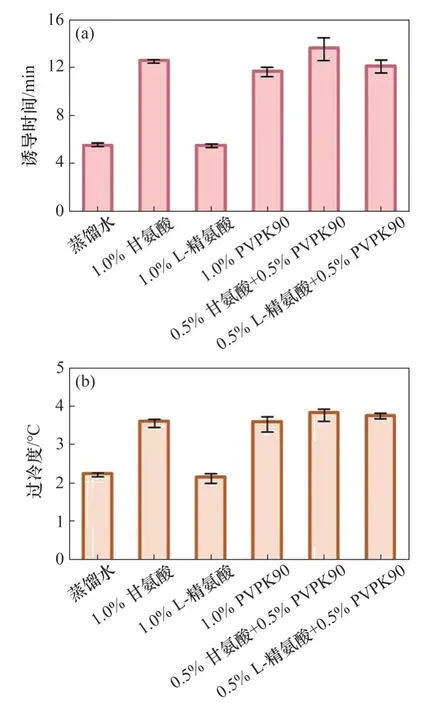
图38 加入相同质量分数的水合物抑制剂时水合物形成诱导时间和过冷度Fig.38 Hydrate induction time and supercooling when the same mass fraction hydrate inhibitor is added
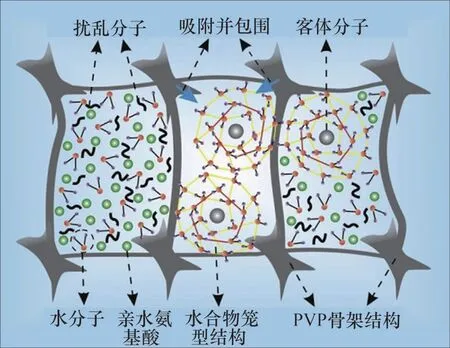
图39 复合体系抑制水合物形成的机理[75]Fig.39 Mechanism of composite system inhibiting the formation of hydrates[75]
2.7.5 多糖
WAN 等[82]研究了5 种多糖(阿拉伯胶、海藻酸钠、瓜尔胶、羧甲基壳聚糖和淀粉)对水合物生成的影响,分子结构如图40所示。从图40可见:多糖并不能抑制水合物成核,但所有多糖都增加了水合物形成的最大过冷度,使得水合物的形成需要更大的过冷度和更高的驱动力,其中瓜尔胶增大水合物形成的最大过冷度能力最强,且其性能优于PVP。经分析认为,多糖的脱水葡萄糖单元具有类似亲水侧链内酰胺基团的特性,能够与水合物笼孔穴结合,阻止气体分子进入,从而抑制水合物的生长。此外,与PVP的内酰胺基团相比,由于多糖中的葡糖酐单元具有羟基,拥有更强的亲水性,能够与水分子形成氢键的能力,因此,多糖的抑制能力更强。
图40 多糖分子结构Fig.40 Molecular structure of polysaccharide
通过研究发现:在不同过冷温度下,多糖表现出不同的抑制性能;在较低的过冷温度(5.0 ℃)下,淀粉显示出最佳抑制性能,但抑制性能随着过冷温度的升高而降低,并且当温度超过其最大过冷温度(7.5 ℃)时,其抑制性能丧失;羧甲基壳聚糖具有类似的特性;而当阿拉伯胶在过冷度为5.0 ℃时,能够在一定程度上抑制水合物的生长,但当过冷温度升高到6 ℃时,它开始促进水合物的形成,并且随着过冷温度升高,促进作用增强。多糖侧链的差异导致其抑制性能不同。瓜尔胶骨架结构上的侧链脱水葡萄糖基团增加了其与水合物表面间的吸附能力,降低了水合物的生长速率;海藻酸钠侧链中羧基的嵌钠化显著提高了其水溶性,从而表现出较强的抑制效果;而阿拉伯胶主要由含少量蛋白质的多糖组成,其侧链以阿拉伯半乳聚糖为主,因此,抑制性能较差。
2.7.6 磺化壳聚糖(SCS)
由于壳聚糖的主要缺点是其溶解度较低,因此,BANERJEE 等[83]将磺酸盐基团引入聚氨酯的主链以增强其溶解性,FARHADIAN等[84]则通过合成磺化壳聚糖(SCS)提高壳聚糖溶解度,其分子结构如图41所示。硫化壳聚糖能够增加水合物形成的时间并降低水合物形成的起始温度,使形成的甲烷水合物显著减少。
图41 硫化壳聚糖分子结构Fig.41 Molecular structure of sulfurized chitosan
SCS的相对分子质量影响其抑制性能,与纯水相比,低相对分子质量的SCS 能够将诱导时间增加3.1~14.3 倍(质量分数为(1~10)×10-3);当存在相对分子质量中等的SCS 时,诱导时间增加了2.1~9.6倍(质量分数为(1~10)×10-3)。
2.7.7 抗冻蛋白(AFP)
MU等[85]通过水合物生成实验比较了昆虫细胞抗冻蛋白mSA-RmAFPI与PVP、组氨酸、赖氨酸、酪氨酸和脯氨酸的动力学抑制性能,结果表明:与所选用的几种氨基酸相比,AFP 能够更有效地抑制水合物成核,将水合物的起始成核温度从11.8 ℃降低到7.8 ℃,与PVP(7.9 ℃)在相同质量分数下的成核效果相同。分析水合物生成过程中的压力变化发现,mSA-RmAFPI降低了水合物的生长速率与水合物产量。另外,较高质量分数的mSA-RmAFPI具有更好的水合物抑制性能。
MADDAH 等[86]通过分子动力学模拟研究了比目鱼AFP I与水合物间的作用,结果表明比目鱼AFPI的存在能够有效降低水合物的形成率。模拟发现AFP I分子中的丙氨酸残基侧链能够进入水合物笼空穴,与水合物笼产生范德华力作用,达到与水合物结合的目的,并且AFP I分子中疏水侧链的疏水作用、水合物表面AFPI侧基与附近水分子之间形成的氢键能够使AFPI稳定在水合物表面。此外,AFPI的亲水表面(含有极性和酸性氨基酸)能够与水形成大部分氢键,非极性丙氨酸与水分子形成较少的氢键,两者在抑制水合物生长方面均起着重要作用。AFP I在水合物/水界面的水合物生长过程中能够发生弯曲现象,当AFPI吸附在水合物表面时,水合物表面围绕AFP 中的氨基酸残基弯曲,水合物表面形成一定曲率,产生了传质阻力,水合物生长受到抑制[87]。
2.8 酯基官能团
由于酯基具有电负性的—C=O—双键,因此,其与酰胺基团类似,能够与水分子相互作用形成氢键,破坏水分子有序结构,抑制水合物成核。此外,酯基还能够与水合物笼形成氢键使抑制剂分子吸附在水合物笼上,从而阻碍水合物进一步生长。酯基基团对水合物的抑制作用如图42所示。
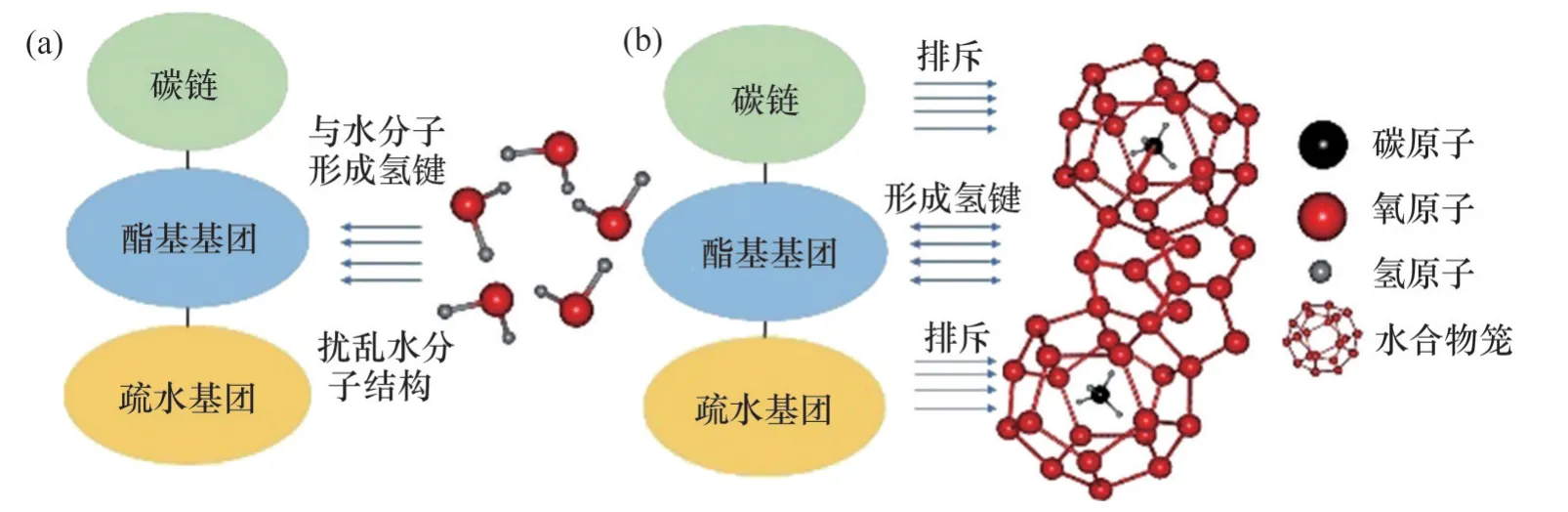
图42 酯基基团对水合物的抑制作用Fig.42 Inhibition of ester groups hydrate
2.8.1 PVCap-b-PCL
乙烯基酯与内酰胺是用于合成动力学抑制剂的良好单体[78],由于两者都具有较强的亲水性,因此,抑制剂能够吸附在水合物表面,减少水分子和水合物笼之间的界面面积,从而阻碍水合物的进一步生长[88]。此外,具有较强疏水性的抑制剂能够干扰水分子结构或者与水合物颗粒表面相互作用[89]。一些两亲性KHIs共聚物(即同时具有亲水性及亲油性)的疏水基团可以调节亲水/疏水平衡,增强共聚物的KHI性能[90],因此,通过对ε-己内酯开环聚合得到具有良好生物降解性、高结晶度和疏水性的聚己内酯链段,而后将其引入到PVCap中,最终获得具有良好生物降解性和抑制性能的两亲性嵌段共聚物PVCap-b-PCL,并且通过适当调整ε-聚己内酯链段的长度,以增强其抑制性能[91]。PVCap-b-PCL 分子结构如图43所示。与PVCap 相比,PVCap-b-PCL 共聚物抑制作用的增强在某种程度上归因于它们在溶液中的胶束化(胶束化是指溶液中表面活性剂的质量分数达到一定值后形成大量分子有序聚集体,其中,表面活性剂分子的疏水基聚集形成胶束内核,亲水的极性基团形成胶束外层)。图44所示为PVCap-b-PCL抑制机制的示意图[88],PVCap-b-PCL的胶束化使得早期水合物形成所需的游离甲烷分子数量减少,并且胶束的疏水性聚己内酯核可以通过范德华力、氢键等方式包裹甲烷分子。此外,由于PVCap-b-PCL 相对分子质量较大,其分子间、分子内链间容易互相缠结,因此,PVCap-b-PCL 不会像小的表面活性剂那样转移到气液界面提高甲烷的溶解度,另一方面,PVCap-b-PCL 分子通过PVCap 片段和水分子之间的相互作用吸附到水合物表面,阻止甲烷分子进入空腔,因此,PVCap-b-PCL 共聚物在水合物生长的早期阶段比PVCap 具有更好的性能。随着水合物的生长,更多的PVCap-b-PCL 分子吸附到水合物表面,并且ε-聚己内酯链将围绕在水合物表面,阻止水合物晶体的继续生长和聚集。
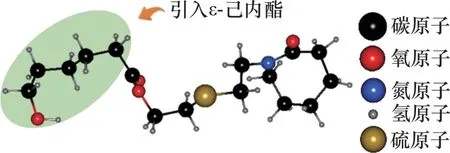
图43 PVCap-b-PCL分子结构Fig.43 Molecular structure of PVCap-b-PCL
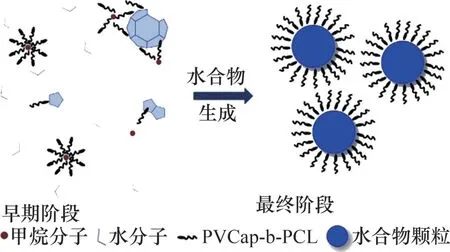
图44 PVCap-b-PCL在甲烷水合物形成中可能的水合物抑制机制示意图[91]Fig.44 Schematic diagram of possible hydrate inhibition mechanism of PVCap-b-PCL in formation of methane hydrate[91]
聚己内酯链段长度影响PVCap-b-PCL 的抑制性能,增加链段长度能够促进胶束化并能增强分子间和分子内的相互作用,抑制性能增强。此外,通过调节可生物降解的聚己内酯链段与PVCap 链段的长度比例,可以获得具有良好生物降解性能的水合物抑制剂。
2.8.2 蓖麻油基聚脲氨酯(CPWUUs)
植物油具有生物降解性,其毒性低,易获得且价格低廉,成为具有潜力的动力学抑制剂。FARHADIAN 等[46]合成了麻油基聚脲氨酯(CPWUUs),并分析了其抑制水合物性能,分子结构如图45所示。结果表明,CPWUUs 中的甲酸酯基、氨基等官能团与水分子相互作用,并且CWPUUs 的脂肪酸链能够使水分子间氢键产生断裂,扰乱水分子氢键网络,达到抑制水合物成核的目的。在水合物生长过程中,CPWUUs 与水合物笼表面的吸附作用阻止了水合物晶体的生长,使水合物生长速率显著降低。此外,由于CPWUUs 蓖麻油结构中存在酯基,因而其具有良好的生物降解性能。
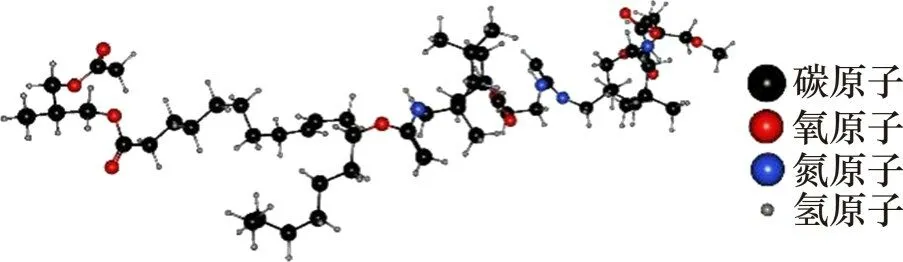
图45 蓖麻油基聚脲氨酯分子结构Fig.45 Molecular structure of castor oil-based polyureaurethane
CPWUUs 的抑制性能受其质量分数影响,随着其质量分数增加,抑制性能逐渐提升。不同相对分子质量的CPWUUs 显示出不同抑制性能。实验结果表明,相对分子质量中等的CWPUUs 具有更好的抑制性能。
2.8.3 P(Vcap-BMA)
DUAN等[92]在PVcap基础上引入了甲基丙烯酸丁酯(BMA)制得P(Vcap-BMA)抑制剂,其分子结构如图46所示。由于引入甲基丙烯酸丁酯的酯基排斥水分子,酰胺环上的氧原子与水分子的相互作用减弱,其疏水性增强。在水中P(VCap-BMA)侧链的疏水基团使其溶解度变低,并且与水合物表面结合形成氢键的能力变弱,因此,水溶液中的P(VCap-BMA)的抑制能力比PVCap 的弱。但当P(VCap-BMA),PVCap 与低剂量甲醇或乙二醇组合使用时,P(VCap-BMA)的溶解度在醇的作用下提高,并且疏水性侧链和更强的空间位阻(指分子中某些原子或基团彼此接近而引起的空间阻碍作用)降低了水相中客体分子与水合物笼的相互作用,从而更有效地抑制水合物,显示出强于PVCap 的抑制性能。
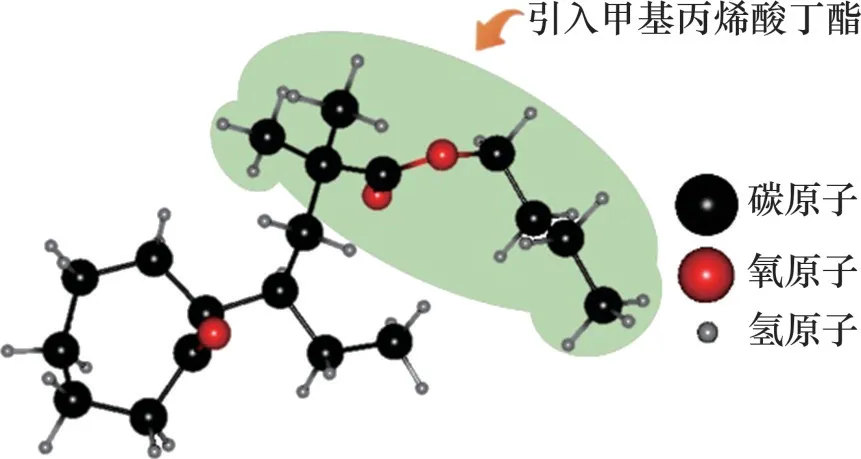
图46 P(Vcap-BMA)分子结构Fig.46 Molecular structure of P(Vcap-BMA)
不同的VCap 和甲基丙烯酸丁酯(BMA)的单体质量比对P(Vcap-BMA)动力学抑制性能存在一定影响,当VCap 与BMA 质量比从3∶1 增加到11∶1时,P(VCap-BMA)中带有酯基的疏水侧链在液态水和水合物核之间的空间位阻效应增强,使得水合物核的生长更加困难,诱导时间增加;而当VCap 与BMA 质量比为13∶1 时,由于疏水基团过多,P(Vcap-BMA)在水中的溶解度降低,抑制作用随之减弱。不同质量分数的P(Vcap-BMA)抑制性能也不同,随着其质量分数增加,P(VCap-BMA)与水合物之间的相互作用增强,使得水分子与水合物表面之间的氢键作用减少,抑制性能增强。
2.9 羧基官能团
位于水合物笼上的水分子中氧原子带负电,相应的氢原子带正电,而羧基中含有2个带正电的氧原子,因此,羧基在水合物表面有3种吸附方式(如图47所示),分别为:羧基中的氢原子与水合物中的氧原子结合;羧基中的氢原子和单键氧原子吸附水合物中的氧原子和氢原子;羧基中的氢原子和双键氧原子被吸附在水合物中的氧原子和氢原子上[64]。羧基通过这3种方式吸附在水合物表面上,达到抑制水合物的目的。
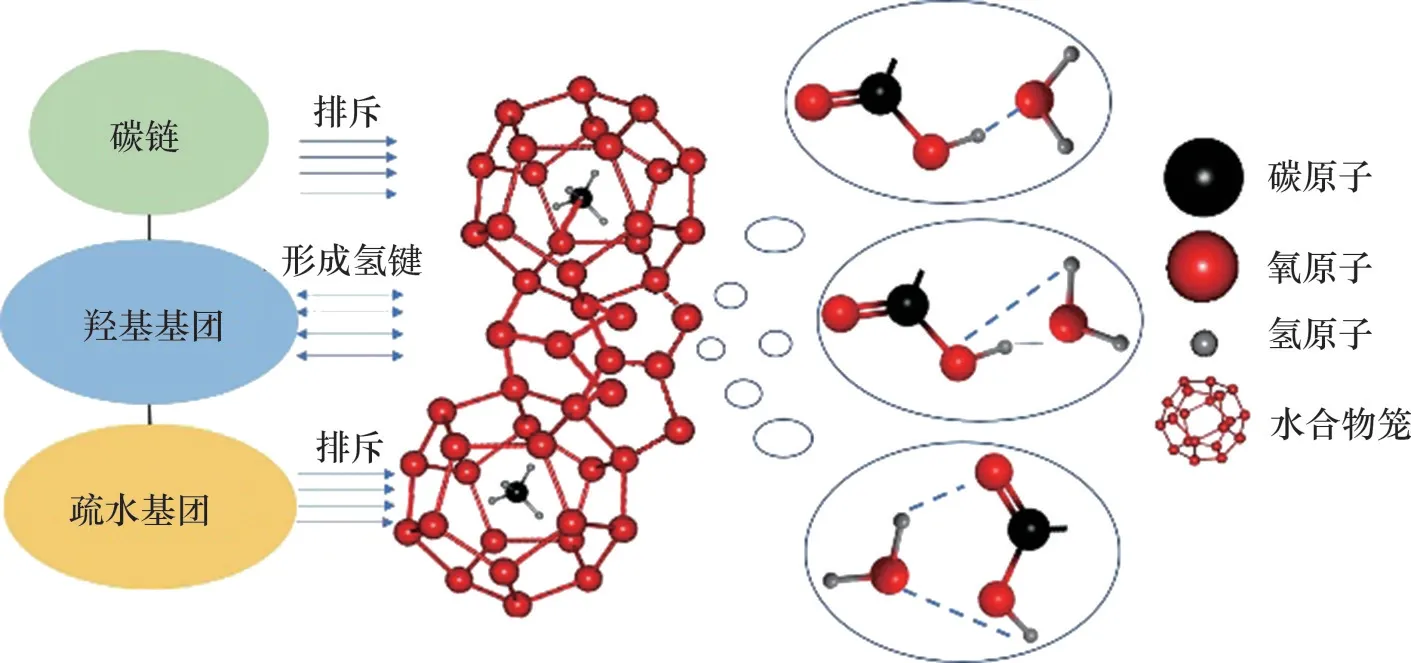
图47 羧基基团对水合物的抑制作用Fig.47 Inhibitory effect of carboxyl groups on hydrates
研究发现一些流动性能良好的防聚剂能够在水-烃界面形成防聚剂层,阻止甲烷从烃相扩散到水合物笼,水合物的生长受到阻碍,表现出动力学抑制性能。
FANG等[93]通过分子动力学模拟了温度与压力分别为260 K和10 MPa时,有无1-苯乙酸,2-萘乙酸和1-芘乙酸存在的水合物生长过程分子结构如图48所示。分析局部水分子状态发现,多环芳香酸的存在减缓了水合物的生长。对多环芳香酸与水合物表面吸附过程进一步模拟发现,多环芳香酸类防聚剂不仅能够通过表面活性剂的疏水性头基(芳香苯环)形成防聚剂薄膜(空间位阻),阻止水合物颗粒聚集,而且可以通过亲水性头基(羧基)与水合物表面结合,抑制水合物生长。其中,具有防聚剂性能的表面活性剂(萘乙酸)能够延迟最高含量为20%(体积分数)水的油系统中水合物生长。
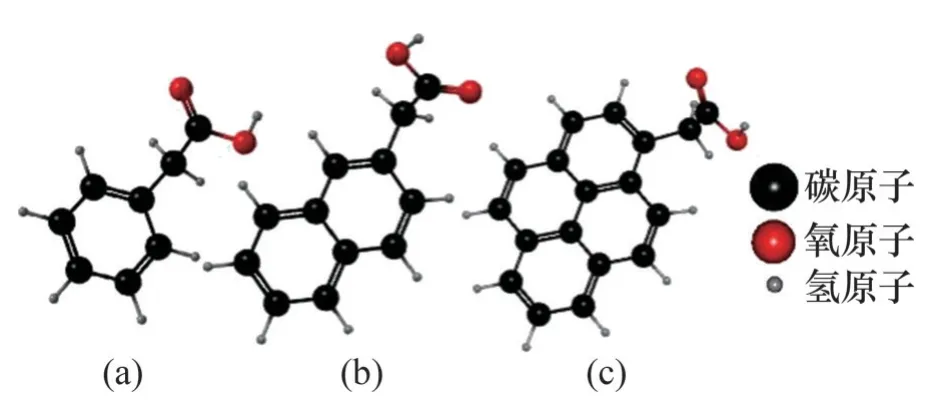
图48 多环芳香酸分子结构Fig.48 Molecular structure of polycyclic aromatic acid
2.10 咪唑基团
2.10.1 1-丁基-3-甲基咪唑四氟硼酸盐、1-丁基-3-甲基咪唑碘
LEE等[94]通过实验与模拟研究了1-丁基-3-甲基咪唑四氟硼酸盐([BMIM][BF4],质量分数为97%)和1-丁基-3-甲基咪唑碘([BMIM][I],质量分数为98%)的动力学抑制性能,分子结构如图49所示,并与甘氨酸、丙氨酸这2种氨基酸进行比较。通过比较水合物生成过程中气体消耗量发现,在3.0 mol%CH4+[BMIM][BF4]作用下,水合物的生长速率远比其他实验组的低。计算水合物笼与抑制剂分子间的相互作用能发现,[BMIM][BF4]比添加甘氨酸具有更高的负相互作用能,[BMIM][BF4]与水合物笼具有更强的相互作用。通过分析拉曼光谱发现,甘氨酸主要抑制51262水合物的形成(51262是一种水合物笼结构,表示由12 个五边形与2 个六边形组成的14 面体),而[BMIM][BF4]抑制512水合物的形成。
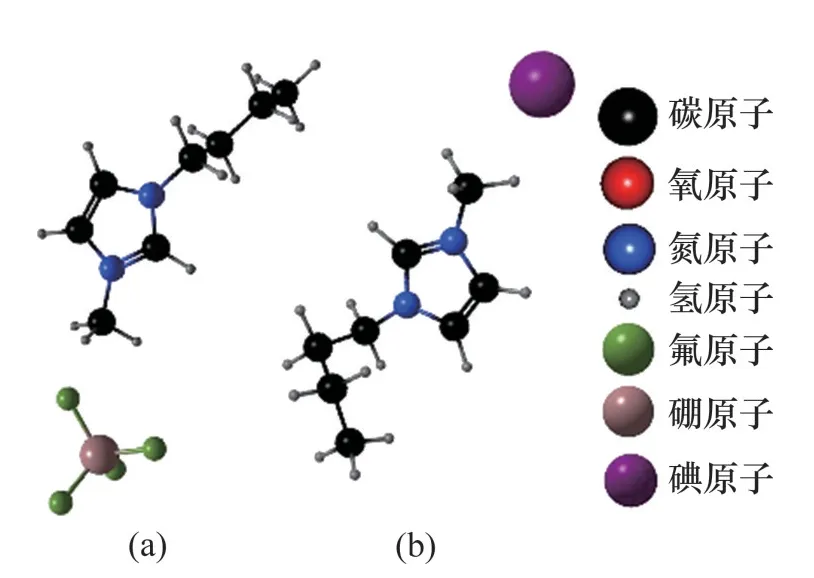
图49 1-丁基-3-甲基咪唑四氟硼酸盐和1-丁基-3-甲基咪唑碘分子结构Fig.49 Molecular structure of 1-butyl-3-methylimidazole tetrafluoroborate and 1-butyl-3-methylimidazole iodine
甘氨酸与[BMIM][BF4]两者抑制水合物存在差异性,使得当两者混合时存在协同抑制的潜力。LEE 等[95]对甘氨酸、PVCa 和[BMIM][BF4]动力学抑制剂两两混合的抑制性能进行研究时,发现在水合物生成的非搅拌实验中,只有甘氨酸(质量分数为0.5%)和[BMIM][BF4](质量分数为0.5%)的混合溶液表现出混合抑制性能,降低了水合物成核起始温度,且其抑制能力比其他抑制剂强。而在搅拌实验中,甘氨酸(质量分数为0.5%)和[BMIM][BF4](质量分数为0.5%)混合溶液,以及PVCap(质量分数为0.5%)和[BMIM][BF4](质量分数为0.5%)混合溶液均表现出协同抑制性能。
2.10.2 甲基咪唑磷酸二氢钠
SULAIMON 等[96]将磷酸二氢钠作为阴离子,合成了3种不同侧链的甲基咪唑磷酸二氢钠,分别为1-乙基-3-甲基咪唑磷酸二氢钠(EMIM DHP),1(3-氰基丙基)-3-甲基咪唑磷酸二氢钠(CPIM DHP)和1-丁基-3-甲基咪唑磷酸二氢钠(BMIM DHP)。甲基咪唑磷酸二氢钠分子结构如图50所示。真实溶剂似导体屏蔽的模拟(COSMO-RS)结果显示:CPMIM阳离子和DHP阴离子可以与水分子中的氢原子形成有效氢键,表明这3种离子液体存在抑制水合物的潜力。通过测量5~15 MPa 压力下,3 种离子液体存在时甲烷水合物形成的诱导时间发现,合成的3 种离子液体抑制剂与PVCap(1.90 h)和Luvicap(0.68 h)相比显示出更好的抑制性能。经分析认为,在水合物成核阶段,离子液体破坏了水和甲烷分子的局部结构,阻碍水合物成核[97]。而在水合物生长阶段,离子液体通过阴阳离子的氢键作用与水合物表面结合,阻碍水合物沿结合生长面进一步生长;此外,离子液体还能够阻止甲烷气体分子进入水合物空穴,并迫使水合物在ILs分子周围或分子间生长[98]。
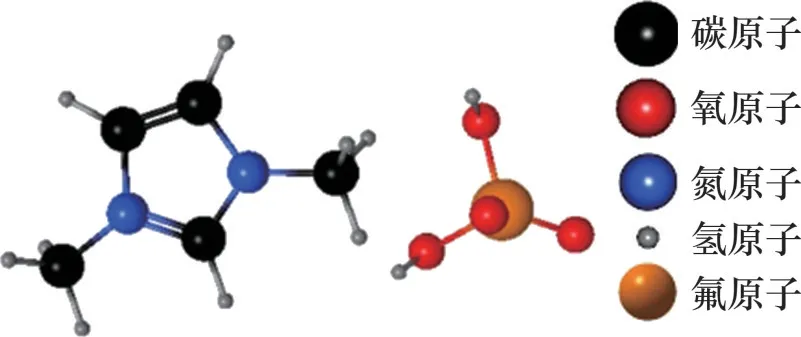
图50 甲基咪唑磷酸二氢钠分子结构Fig.50 Molecular structure of methylimidazole sodium dihydrogen phosphate
在不同压力下,离子液体显示出不同的抑制性能,但总体来说,在压力较低时,诱导时间较长。其中,EMIM DHP 在实验压力范围内,诱导时间随着压力增加而减少。先前研究表明,在抑制甲烷气体水合物的成核和生长方面,相对分子质量较大的离子液体具有较好的性能,因为它能够产生空间位阻,使水和甲烷气体分子彼此远离,并且更高的分子间作用力将导致ILs和水合物表面之间的相互作用更强[99]。但3种离子液体的平均诱导时间随烷基链长的增加而缩短。经分析认为,由于阳离子和阴离子体积都很大,烷基链长度增加使离子液体分子,导致3种离子液体的抑制性能下降。
3 结论与展望
1)多数动力学抑制剂为表面活性剂,抑制剂分子中疏水基团的疏水作用能够使水分子在其周围形成水笼,使得聚合物能够与水合物空腔结合,产生位阻效应,也能使水分子间氢键断裂,扰乱水分子氢键网络。而亲水基团可以通过与水分子产生氢键阻止水合物笼的形成,并且也能够吸附在水分子笼上,阻碍客体分子进入水合物空穴,而较小的亲水基团还可以占据水合物空穴。
2)寻找并引入性能优异的官能团或特定分子链段、对动力学抑制剂进行分子设计、调整聚合物单体比例等是目前研发动力学抑制剂的重要技术手段。
3)水合物的抑制机理具有差异性或在不同阶段对水合物具有抑制能力的动力学抑制剂之间(如L-酪氨酸、PVP 之间等)存在着协同抑制作用,该现象较普遍。
4)动力学抑制性能普遍受动力学抑制剂质量分数、相对分子质量、聚合物单体质量比、所处环境温度的影响,其中部分动力学抑制剂如PVP-A、淀粉、聚乙二醇等在不同环境温度或不同相对分子质量时,会促进或抑制水合物生成。
5)动力学抑制剂的分子构象对抑制剂能否充分发挥抑制性能具有显著影响。动力学抑制剂的分子构象决定了动力学抑制剂能否充分伸展,能使抑制剂分子与水分子、水合物笼充分接触,充分发挥其抑制能力。
6)部分动力学抑制剂能够从不同方面抑制水合物的生成,如聚乙二醇、多环芳香酸,聚乙二醇不仅具有动力学抑制性能,而且是一种代表性热力学水合物抑制剂,而多环芳香酸是一种性能良好的防聚剂。
7)动力学抑制剂的研发应以工程实际为基础,开发的动力学抑制剂应当具有性能优异、成本低、污染低、毒性低等特点。但目前已开发的动力学抑制剂如阿拉伯胶、PVP 和PVP-A 等在过冷度较高时抑制能力丧失,反而促进水合物的生长,并且酰胺类等非天然动力学抑制剂普遍存在生物降解性不足的缺陷,如何解决这些问题是未来需要思考的问题。
8)目前的各类改性抑制剂如改性PVCap、改性PVA 等仅以提高浊点或抑制能力等单一目的进行化学改性,并没有进行多方位综合考虑,因此,可以分析现有抑制性能较好的动力学抑制剂存在的缺陷,综合考虑浊点、过冷度、生物降解性等,引入2个或多个官能团或分子链获得综合性能优秀的动力学抑制剂。
9)抑制剂分子的构象影响其性能的发挥,如何控制分子构象使得动力学抑制剂分子充分伸展,使动力学抑制剂与水合物、水分子充分接触是未来研究动力学抑制剂时需要思考的问题。
10)不同动力学抑制剂之间普遍存在协同抑制性能,因此,可以分析2种或几种分别在水合物成核与生长的不同阶段具有优秀抑制性能并存在协同抑制效应的动力学抑制剂,以发挥协同抑制效应产生更强的抑制能力。另外,混合的抑制剂质量分数与混合比例是否对动力学抑制剂混合溶液的协同抑制作用与抑制水合物能力存在影响尚不明确,有待进行实验研究。
11)目前,对油水两相体系中水合物抑制剂的研究主要集中在防聚剂,而动力学抑制剂的研究主要集中在水相,但油水两相体系是否影响动力学抑制剂的抑制行为尚不明确。
12)目前,对于动力学抑制剂的微观机理仍不明确,需进一步研究。
