基于密度泛函理论的水对黄铁矿和煤表面润湿性机理研究
2022-03-30王成勇陈鹏谭金龙方永城
王成勇 ,陈鹏 ,谭金龙 ,方永城
(1. 六盘水师范学院 化学与材料工程学院, 贵州 六盘水 553004;2. 中国矿业大学化工学院, 江苏 徐州 221116;3. 中国矿业大学(北京) 化学与环境工程学院, 北京 100083)
浮选是矿物分选的主要方法之一,其建立在矿物表面润湿性差异之上[1]。矿物表面对水的润湿性大小由表面吸附水分子的难易程度所决定。亲水性矿物表面易吸附水分子形成水化膜,阻碍矿物颗粒与气泡的附着;疏水性矿物表面不易形成水化膜或水化膜较薄不稳定,矿粒与气泡接近时,表面张力促使水化膜薄化破裂,最终矿粒附着于气泡上,矿化气泡上浮形成泡沫产品[2]。因此,对矿物表面润湿性机理进行研究,可以为矿物浮选过程中表面润湿性调控提供理论依据。
随着量子化学计算的发展,基于密度泛函理论(DFT)的量子化学计算方法被广泛用于矿物表面润湿性研究。Stirling等采用DFT理论分析了水分子在黄铁矿(100)表面的吸附,发现水分子主要吸附在表面Fe原子上[3]。陈建华等采用DFT研究了硫化矿表面的润湿性,水对黄铁矿和闪锌矿的润湿性强于对方铅矿和辉钼矿的润湿性[4]。高正阳等在采用DFT分析不同煤阶煤表面的润湿性时,发现4种不同煤阶煤吸附水分子的相互作用由大到小的顺序为:褐煤>次烟煤>烟煤>无烟煤[5]。目前,学者们在分析水对矿物表面润湿性机理时主要考虑水分子在矿物表面的吸附情况,而润湿过程中气-水分子的竞争吸附鲜有报道。本文采用Materials Studio软件的CASTEP模块进行DFT计算,分析了黄铁矿和理想化的煤表面的弛豫和电子态密度,以及水分子在其表面上的吸附和气-水分子竞争吸附。
1 计算方法与模型
理想的黄铁矿为均一物质,具有周期性的晶体结构,而煤的结构比较复杂,为了计算和分析简便,需对煤的结构进行合理简化。随着煤化程度增加,煤的C含量增高而杂原子含量减少,煤的芳香环数增加,逐渐向石墨结构转变,因此可以利用石墨结构作为煤的理想化结构模型,煤中杂原子、侧链和官能团本文不做考虑,可在后续研究中当作理想化的煤表面的缺陷进行研究[6]。浮选过程中气泡的气体为空气,空气的主要成分为氮气和氧气,分别约占空气的78%和21%;其中氮气分子具有很好的对称性,且化学性质稳定,在常温常压下,很难与矿物表面发生吸附或化学反应,因此研究空气在矿物表面上的吸附以氧气为例。
采用密度泛函理论(DFT)对表面模型和吸附构型进行几何优化和性质分析。从体相中切出表面模型,再与水分子、氧分子构建吸附构型。黄铁矿表面、煤表面、水分子和氧分子模型见图1。黄铁矿(100)表面含有6层原子,理想化的煤表面含有2层原子,表面上的真空层厚度均为8Å[7]。体相、表面和吸附构型的几何优化条件为:交换-相关泛函采用广义梯度近似(GGA)框架下的PBE泛函,几何优化算法采用准牛顿算法(BFGS),价电子和离子实的相互作用采用超软赝势(USP),不考虑自旋极化修正,截断能为300 eV,能量收敛标准为2.0×10-5eV/atom,晶体内应力收敛标准为0.05 GPa,原子位移收敛标准为0.002 Å,自洽迭代收敛精度为2.0×10-6eV/atom。原子间作用力收敛标准:体相为0.03 eV/Å,表面和吸附构型为0.05 eV/Å;Brillouin区积分计算:体相采用6×6×6,表面采用5×5×2,吸附构型采用4×4×2[8]。单个水分子和氧分子均置于15×15×15 Å周期性晶胞中进行几何优化,优化后水分子键角和键长为:∠H-O-H=104.649°,dO-H=0.975 Å,氧分子的键长为1.242 Å,这与参考文献中的结构参数相近[9]。
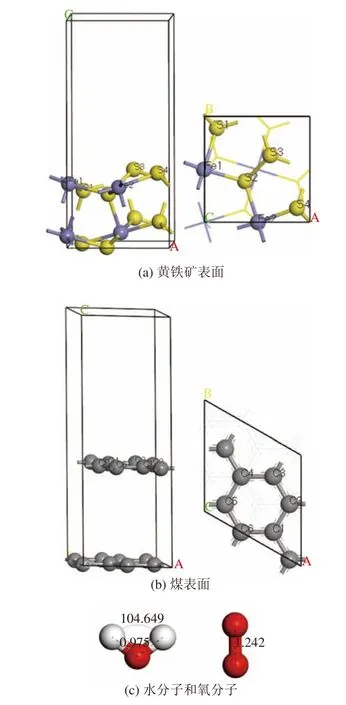
图1 黄铁矿表面、煤表面、水分子和氧分子模型Fig.1 Models of pyrite surface, coal surface, water molecule and oxygen molecule
吸附能计算公式为:

式中:Einteraction表示吸附质吸附在表面上的吸附能,kJ/mol;E(surf+ads)表示吸附质分子吸附在表面后整个体系的总能量,kJ/mol;Esurface表示表面的能量,kJ/mol;Eads表示吸附质分子的能量,kJ/mol。吸附能为负值,表明吸附为自发过程,且值越小吸附越易进行[10];吸附能为正值,表明吸附为吸热过程,值越大吸附越难进行。
2 结果与讨论
2.1 表面弛豫
当矿物沿某一方向产生表面时,表面原子会发生弛豫,黄铁矿和煤表面原子弛豫见表1。由于表面上方为真空,Fe1和Fe2原子缺少Fe-S键的作用,导致往z轴负方向移动;S3和S4为顶层原子,且缺少一个配位,因此向z轴负方向移动;而S1和S2为第三层原子,原子位移很小,几乎未发生弛豫。煤表面原子发生了轻度弛豫,表面上方没有原子,表面第一层原子只受到下层原子的吸引作用,使得表面原子往z轴负方向移动。黄铁矿表面原子与体相原子相比,原子的配位数发生了改变,Fe原子为5配位,S原子为3配位和4配位,因此Fe1、Fe2、S3和S4有未成键的悬挂键;而煤表面原子配位数没有改变。因此,可以预见黄铁矿表面原子具有更强的活性,更易与吸附质(水分子和氧分子)发生作用。

表1 黄铁矿和煤表面原子位移和配位数Table 1 Atomic displacement and coordination on the surface of pyrite and coal
2.2 表面态密度
态密度(Density of State,DOS)代表能量间隔内的量子态数,反应了电子的分布;低能级处的态密度代表了电子相对较稳定,高能级处的态密度则代表了电子相对较活跃;费米能级是电子全空和全满的标志,费米能级附近的电子最活跃,电子首先在此处发生转移[11]。
黄铁矿和煤表面的态密度见图2,黄铁矿表面高能级处态密度分布较多,低能级处分布较少,且在费米能级附近出现较高峰,说明黄铁矿表面电子比较活跃,吸附活性较强;而煤表面低能级处态密度较多,高能级处态密度较少,且费米能级附近态密度较少,说明煤表面对电子的束缚较强,吸附活性较弱。
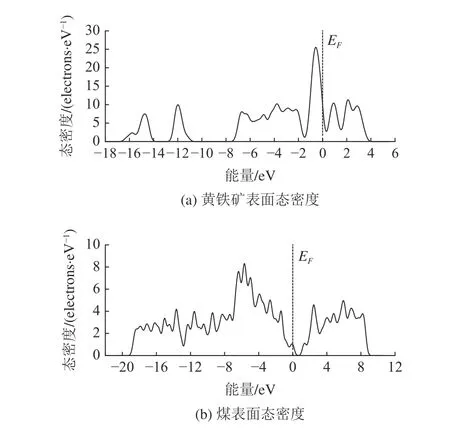
图2 黄铁矿和煤表面态密度Fig.2 DOS of pyrite and coal surfaces
2.3 水分子在黄铁矿和煤表面上的吸附
由于硫原子电负性大于铁原子,水分子中氧原子电负性大于氢原子,因此氢原子易与硫原子发生相互作用,氧原子易与铁原子发生相互作用[12]。水分子在黄铁矿表面上的吸附构型考虑四种:(a)硫顶位,水分子的一个氢原子对着第一层的硫原子S3;(b)铁顶位,水分子氧原子对着第二层的铁原子Fe1;(c)底部对硫穴位,水分子处于穴位上,两个氢原子分别对着第三层的硫原子S1和S2;(d)顶部对硫穴位,水分子处于穴位上,两个氢原子分别对着第一层的两个硫原子S3和S4。平衡吸附构型见图3。

图3 水分子在黄铁矿表面上的平衡吸附构型Fig.3 Equilibrium adsorption configuration of water molecule on pyrite surface
设计煤表面吸附构型时,水分子的两个氢原子向下[13]。水分子在煤表面的吸附构型考虑2种:(a)对碳穴位,水分子位于芳香环中心,两个氢原子向下对着碳原子C4和C1;(b)碳顶位,水分子在碳原子C4上方,两个氢原子向下,平衡吸附构型见图4。
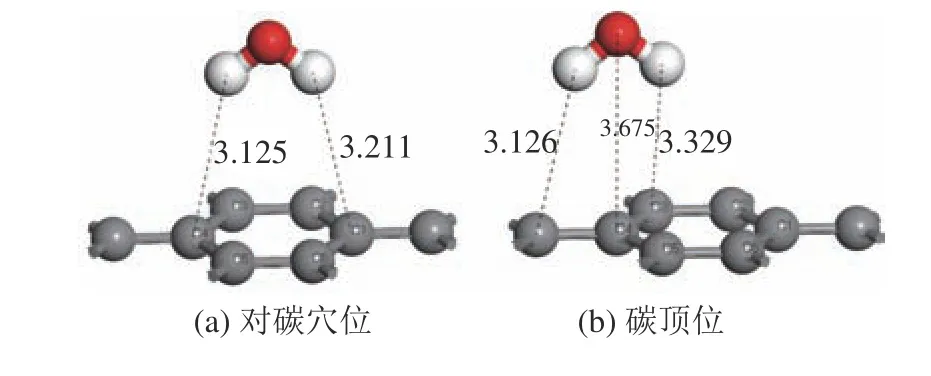
图4 水分子在煤表面上的平衡吸附构型Fig.4 Equilibrium adsorption configuration of water molecule on coal surface
水分子在黄铁矿和煤表面上的吸附能见表2。水分子在黄铁矿表面上各吸附位的吸附能均为负,说明水分子易吸附在黄铁矿表面上。其中底部对硫穴位(见图3c)的吸附能最负,为-87.42 kJ/mol,远小于其他几种吸附构型,这表明该吸附构型最稳定,水分子最容易以这种方式吸附到黄铁矿表面上;其次为铁顶位(见图3b),其吸附能为-38.79 kJ/mol;而硫顶位(见图3a)和顶部对硫穴位(见图3d)的吸附能较大,水分子的吸附不易进行。水分子在煤表面的吸附能均为正值,说明水分子难以吸附于煤表面上。

表2 水分子在黄铁矿和煤表面上的吸附能Table 2 Adsorption energy of water molecule on pyrite and coal
最稳定吸附构型的电子密度分析结果如图5所示。从图5(a)可知,水分子以底部对硫穴位的方式吸附在黄铁矿表面上时,水分子与黄铁矿表面存在电子分布,说明水分子与黄铁矿表面原子之间相互作用较强。从图5(b)可知,水分子与煤表面之间没有电子分布,说明水分子不易吸附到煤表面上。综上所述,黄铁矿表面具有较强的亲水性,而煤表面具有较强的疏水性。

图5 黄铁矿和煤表面吸附水分子的电子密度Fig.5 Electron density of adsorbed water molecules on the surface of pyrite and coal
2.4 氧分子在已吸附水分子表面上的吸附
以水分子在黄铁矿表面的底部对硫穴位和在煤表面的对碳穴位吸附构型为初始吸附模型,再进行氧分子的竞争吸附分析。氧分子在已吸附水分子的黄铁矿表面的吸附位选取3种:(a)底部对硫穴位,两个氧原子分别对着S1和S2;(b)平行硫铁键,两个氧原子分别对着S3和Fe2;(c)对铁穴位,氧分子平行于穴位,两个氧原子分别对着Fe1和Fe2。平衡吸附构型见图6。
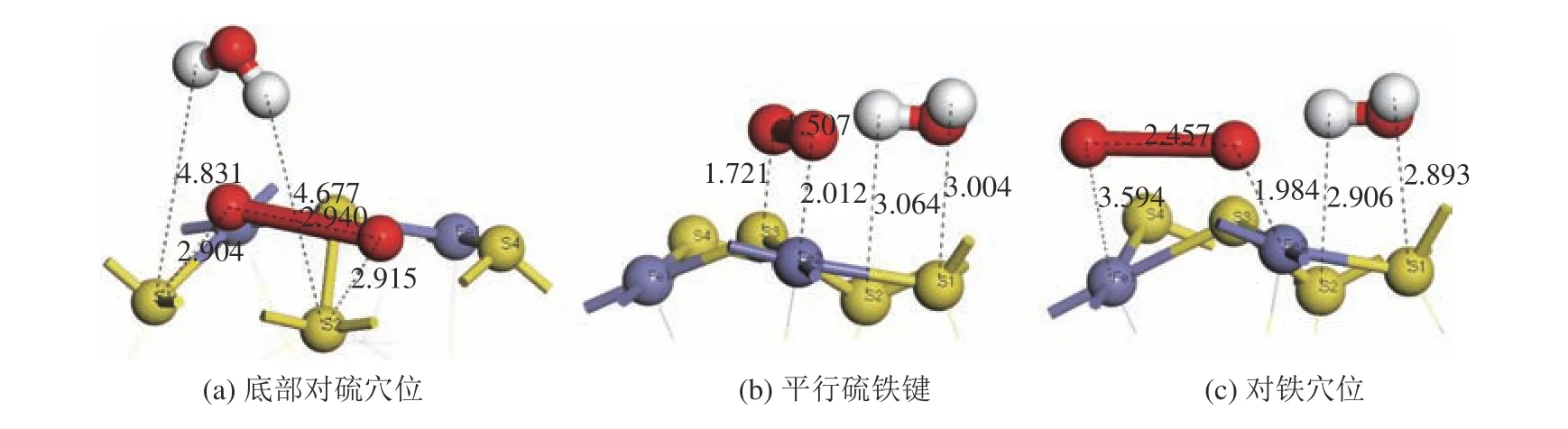
图6 氧气分子在已吸附水分子的黄铁矿表面的平衡吸附构型Fig.6 Equilibrium adsorption configuration of oxygen molecule on the pyrite surface adsorbed by water molecule
氧分子在已吸附水分子煤表面的吸附构型选取3种:(a)邻碳位,两个氧原子分别对着C3和C4;(b)间碳穴位,两个氧原子分别对着C2和C4;(c)对碳穴位,两个氧原子分别对着C1和C4。平衡吸附构型如图7所示。

图7 氧气分子在已吸附水分子煤表面的平衡吸附构型Fig.7 Equilibrium adsorption configuration of oxygen molecule on coal surface adsorbed by water molecule
氧分子在已吸附水分子的煤表面上的吸附能见表3。氧分子在已吸附水分子的黄铁矿表面上的吸附能均为负值。其中对铁穴位(见图6c)的吸附能最小,为-499.31 kJ/mol,明显小于其他两种吸附构型,这种吸附方式最稳定,氧气分子最易以这种方式在黄铁矿表面吸附;同时氧分子的两个氧原子发生了解离,氧原子分别与铁原子发生相互作用,使黄铁矿表面发生氧化;与图3(c)比较,吸附于表面上的水分子与黄铁矿表面的距离只发生了微小的变化,可以认为氧分子的介入对水分子在黄铁矿表面的吸附影响较小。其次为底部对硫穴位(见图6a),氧分子吸附于黄铁矿表面上且发生了解离,同时水分子被排开。平行硫铁键(见图6b)的吸附能略大于底部对硫穴位,氧分子未发生解离,水分子与黄铁矿表面之间距离几乎未发生变化。

表3 氧分子在已吸附水分子的矿物表面上的吸附能Table 3 Adsorption energy of oxygen molecule on the mineral surface adsorbed by water molecule
氧分子在已吸附水分子煤表面的吸附能:邻碳位(见图7a)的吸附能为正值,与图4(a)比较,水分子的位置没有发生大的变化,只有对着C4的氢原子一端略微抬起;因此这种吸附方式不易发生。间碳穴位(见图7b)和对碳穴位(见图7c)的吸附能均为负值,氧分子容易发生吸附;水分子与煤表面之间的距离变得很大,说明氧分子的介入会将水分子排离煤表面;其中间碳穴位的吸附能最小,为-26.75 kJ/mol,说明在已吸附水分子的煤表面,氧分子最易在间碳穴位发生吸附,并且排开水分子。
最稳定吸附构型的电子密度分析结果如图8所示。从图8(a)可知,氧分子与黄铁矿表面之间存在电子分布,说明氧分子与黄铁矿表面之间相互作用较强;而水分子与黄铁矿表面之间仍然有电子分布,说明氧分子的吸附对水分子的吸附影响较小。从图8(b)可知,水分子、氧分子和煤表面相互之间都没有电子分布,说明三者之间相互作用较弱。

图8 氧气-水分子竞争吸附的电子密度Fig.8 Electron density of oxygen-water competitive adsorption
3 结论
(1)黄铁矿(100)表面发生了明显的驰豫,Fe和S原子均有未成键的悬挂键,且表面电子性质活跃,使得黄铁矿表面具有较强的吸附活性;而理想化的煤表面原子只发生了轻度驰豫,表面原子配位数与体相相同,且表面对电子的束缚较强,使得理想化的煤表面吸附活性较弱。
(2)黄铁矿表面具有较强的亲水性。水分子在黄铁矿表面上各吸附位的吸附能均为负值,在底部对硫穴位的吸附最稳定(吸附能为-87.42 kJ/mol),且与表面之间存在电子分布,表明水分子易吸附在黄铁矿表面上;氧分子易吸附于对铁穴位,并发生解离,与表面之间存在电子分布,但其对水分子的吸附影响较小。
(3)理想化的煤表面具有较强的疏水性。水分子在理想化的煤表面上各吸附位的吸附能均为正值,且与表面间没有电子分布,表明水分子不易吸附在理想化的煤表面上;氧分子在理想化的煤表面上吸附时未发生解离,在间碳位和对碳位的吸附能为负值,并将水分子排离表面。
