基于环丁烯-1,2-二羧酸分子的二维有机铁电分子晶体单层的设计与理论研究*
2022-03-30童健马亮
童健 马亮
(东南大学物理学院,南京 211189)
1 引言
自石墨烯被成功制备以来[1],二维材料因具有独特和优良的性质而受到了极为广泛的关注和研究[2-7].特别地,具有某些功能性的物理性质,如铁电、铁磁等性质的二维功能材料,将有望在微电子、光电和能源转化等应用器件的小型化、微缩化、快速、高灵敏和低功耗等方面发挥巨大的潜能.这极大地激发了人们对二维铁电、铁磁等功能材料的研究热情[8-11].事实上,人们在铁电材料准二维超薄膜的制备和研究方面进行了长期的努力[12,13].然而,由于在准二维铁电超薄膜中退极化场得到了显著增强,从而导致铁电超薄膜的自发极化普遍较低[14],亦使得超薄膜材料的铁电畴稳定性被严重削弱.因此铁电材料准二维超薄膜的研究虽历时已久,却鲜有突破性进展.近年来,二维铁电领域迎来了重大转机,在原子厚度的二维铁电薄膜和新型二维铁电材料的研究方面取得了突破性成果.一些铁电薄膜,如PbTiO3,BaTiO3和BiFeO3等,已陆续在实验上被合成[15-18].实验证实了原子厚度的二维双元化合物,如SnTe 和WTe2,分别具有二维面内和垂直于二维平面的自发极化[19,20].第IV主族的硫族化合物[21,22](如SnS,SnSe,GeS,GeSe)和第V 主族的二维二元合金材料[23](如SbN,BiP)也被第一性原理计算预测具有铁电性.自从具有面外自发极化的CuInP2S6(CIPS)二维单层被实验合成以来[24],相关的理论计算预测了与该结构相似的二维材料,如单层CuCrP2S6和AgBiP2Se6,亦具有面外铁电性[25-27].Ding 等[28]通过第一性原理计算预测了一些由第III 主族和第VI 主族元素组成的二元化合物的单层结构具有铁电性,其中In2Se3薄膜已在实验中被成功合成[29].类似地,理论计算指出一些铋的氧硫族化合物的单层结构也是具有优良半导体性质的铁电材料[30].近年来,原子厚度的NaOH 和CrOOH 薄膜的氢键铁电性及其独特的质子转移机制也被第一性原理计算所证实[31,32].
自从1921 年罗息盐的铁电性被证实以来[33],人们对有机铁电分子晶体及其物理机理开始有了逐步深入的认识和理解.相比于无机体系而言,有机铁电分子晶体往往具有结构及化学多样性丰富、质量轻、柔性好、不含重金属原子及成本低和易于大规模制备等优势[34-36],在最近20 年受到越来越多的关注和研究.但二维有机铁电分子晶体却鲜有报道.近年来,随着二维材料的研究与发展,陆续有理论提出将二维无机材料骨架与极性的有机官能团相结合,是一种在二维体系中引入极化特性的替代方案,并且这一思路在实验上取得了一定的进展[37-41].例如,实验中合成了具有强自发极化的二维有机无机杂化铁电材料[Hdabco][ReO4],其居里温度达到了499 K 且具有极快的铁电反转速率[37];通过向二维钙钛矿框架中引入氟化的芳香阳离子,实验中合成了二维杂化铁电体[2-FBA]2PbCl4[40].上述二维铁电材料本质上是在二维无机骨架上结合有机的极性功能团而实现的,并没有充分体现有机铁电材料的优势.为此,迫切需要寻求新型二维全有机铁电分子晶体,而在此方面的研究较为缺乏,最近,Ma 等[41]通过第一性原理计算成功预测了一种基于机械/化学剥离的二维全有机铁电材料—二维DIPAB (diisopropylammonium bromide),这项工作为实现二维全有机铁电材料提供了新的思路.受此启发,本文对数种基于单一组分的二维质子转移性有机铁电分子晶体进行研究,通过第一性原理计算提出了一种基于环丁烯—1,2-二羧酸(cyclobutene-1,2-dicarboxylic acid,CBDC)的新型二维单层有机铁电分子晶体CBDC (102),它可由沿着CBDC 分子晶体块体的(102)晶面进行剥离而获得,其高自发极化值和良好的铁电稳定性亦被第一性原理计算所证实.
2 计算方法
本文主要采用基于密度泛函理论的第一性原理计算方法,使用软件VASP (Viennaab-initiosimulation package,版本5.4)[42]对材料的铁电相关性质进行模拟计算.计算采用广义梯度近似[43]下的Perdew-Burke-Ernzerhof[44]泛函,离子实和价电子间的相互作用采用投影缀加平面波[45]来描述.在结构优化中采用了DFT-D3 方法[46]来修正分子间的范德瓦耳斯相互作用.在计算中,平面波截断能设置为450 eV,能量收敛精度和力收敛的精度分别设置为10—5eV 和0.01 eV·Å—1.采用Γ中心的Monkhorst-Pack 方法进行第一布里渊区采样[47],对块体进行晶格优化时,选取了7 × 3 × 5的k点网格;对单层结构进行优化时k点网格的选取为3 × 3 × 1.针对二维结构的平板模型,在z方向设置了厚度超过12 Å的真空层以避免周期性边界条件所导致的相邻层间的相互作用.采用贝里相位(Berry phase)方法[48]对材料的铁电极化性质进行了模拟计算.为了确定由于周期性边界条件而引入的极化量子,以相应的顺电相作为参考相(自发极化Ps=0),建立一系列铁电相和参考相之间的中间结构,并以质子位移为序参量(λ),计算得到极化值与质子位移间的连续变化曲线,通过与顺电相的对比确定极化量子.材料铁电极化反转势垒的计算采用线性插入中间结构的方式,从始末结构出发构建出多个中间结构并计算该结构与初始结构的能量之差从而确定反转势垒.
3 结果与讨论
Horiuchi 等[49-52]近年来陆续发现并合成了多种基于质子互变异构的全有机铁电材料.我们观察到其中的CBDC,3-HPLN 和PhMDA 几种分子晶体的铁电性来源于晶体中分子的氢原子和氧原子之间的氢键O—H···O 的定向排列,这种氢键的定向排列形成了极性的氢键链,从而使得材料体现出宏观铁电极化.为了探究这些材料是否有可能通过对相应的块体沿指定晶面进行剥离而制备,对这些块体的结构进行了细致观察,相应的材料结构如图1—图3 所示.观察结构图可以发现,在PhMDA或HPLN 分子晶体中并不存在界限清晰且定义明确的单层结构,然而在CBDC 分子晶体中,CBDC分子自发有序地排列在晶体的(102)晶面上从而形成界线清晰的单层结构,块体可视为这些单层结构在垂直于分子平面的方向上堆叠的产物.
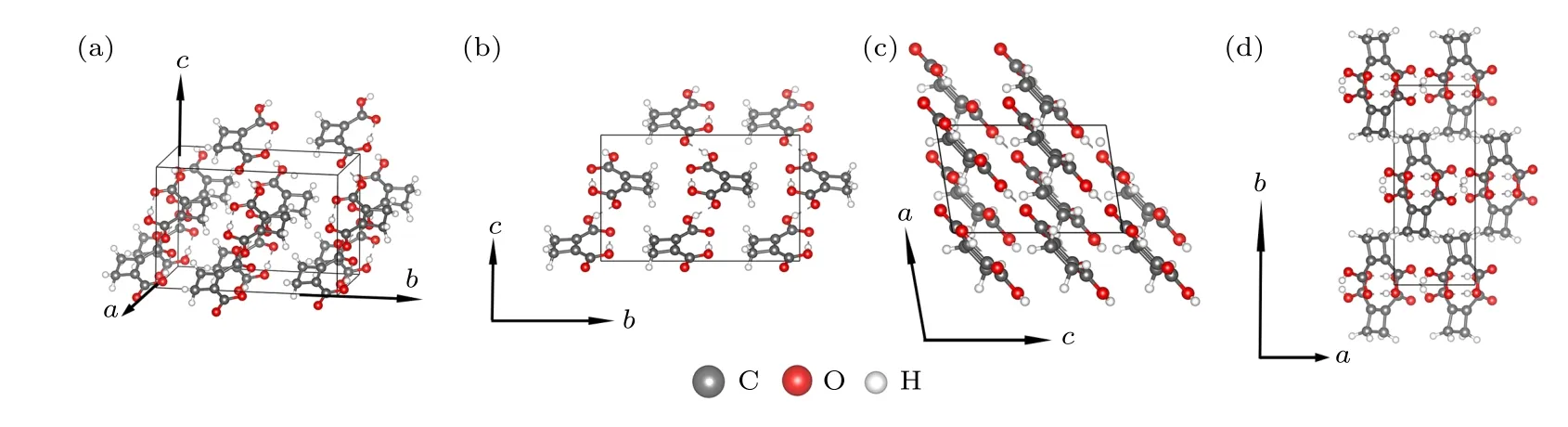
图1 优化后的CBDC 分子晶体结构图示 (a) CBDC 块体的斜视图;(b)—(d)分别为晶胞沿a,b,c 基矢方向的视图.图中灰色、白色和红色小球分别指代碳原子、氢原子和氧原子Fig.1.Optimized structure of bulk of CBDC molecular crystal:(a) Perspective view of bulk CBDC;(b)—(d) view of bulk CBDC along a,b and c axis,respectively.The gray,white and red balls denote C,H and O atoms,respectively.

图2 优化后的块体3-HPLN 分子沿晶胞不同基矢方向的晶体结构视图 (a) 基矢a 方向;(b) 基矢b 方向;(c) 基矢c 方向.图中灰色、乳白色和红色小球分别指代碳原子、氢原子和氧原子Fig.2.View of optimized structure of bulk 3-HPLN molecular crystal along different axes:(a) Along a axis;(b) along b axis;(c) along c axis.The gray,milk white and red balls denote C,H and O atoms,respectively.
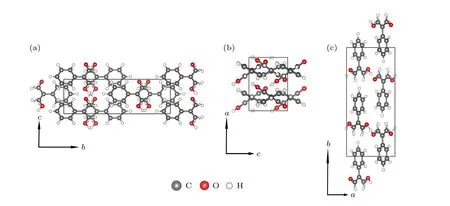
图3 优化后的块体PhMDA 分子沿晶胞不同基矢方向的晶体结构视图 (a) 基矢a 方向;(b) 基矢b 方向;(c) 基矢c 方向.图中灰色、乳白色和红色小球分别指代碳原子、氢原子和氧原子Fig.3.View of optimized structure of bulk PhMDA molecular crystal along different axes:.(a) Along a axis;(b) along b axis;(c) along c axis.The gray,milk white and red balls denote C,H and O atoms respectively.
CBDC 块体的铁电极化物理机理如图4 所示.CBDC 分子内部的氢原子和氧原子之间形成分子内氢键,同时,相邻分子间的氢原子和氧原子结合形成分子间氢键,这两种氢键交替排列形成定向延伸的氢键链,由于氢键本身具备极性特质,使得材料出现平行于氢键链方向的宏观自发极化.从图4可以看出,当材料的铁电极化发生反转时其中的质子发生了集体迁移,即C—O—H···O=C 结构转变为C=O···H—O—C,反之亦然.
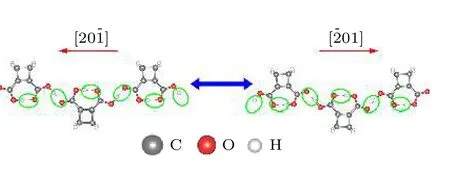
图4 CBDC 晶体的铁电机制.图中绿色圆圈标注了氢键的位置,其中质子的集体迁移导致了铁电极化由晶格[20¯1]方向转为 [ ¯201] 方向;灰色、乳白色和红色小球分别指代碳原子、氢原子和氧原子Fig.4.Ferroelectric mechanism of CBDC crystal.Green circles mark the collective transfer of protons within the hydrogen bonds,leading to the orientation of polarity being reversed from [ 20¯1] direction to [ ¯201] direction.The gray,milk white and red balls denote C,H and O atoms,respectively.
初始的CBDC 块体结构来源于实验中的X 射线衍射数据[50],因此结构中氢原子的位置具有较大的不确定性.为了消除上述氢原子位置的误差,使得初始结构更加符合实际,文中对CBDC块体进行结构优化时,将其中氢原子以外的其他原子进行位置固定而仅让氢原子在力场作用下移动.块体CBDC 自发极化的实验结果和理论值如表1 所列,计算结果显示CBDC 块体的自发极化值为15.0 µC/cm2,其中11.7 µC/cm2沿着基矢a方向而9.5 µC/cm2垂直于该方向.值得注意的是,对于铁电相下的CBDC 块体,沿着基矢a方向或c方向的自发极化无法通过实验单独测量得出[52],因此其实验值和理论值之间有不可忽视的偏差,本文遵循先前文献中的处理方法,即将Horiuchi 等[52]的计算结果作为参考值.计算所得的自发极化与参考值符合得很好,意味着本文构建的CBDC 块体模型以及相应的铁电极化计算是合理的.
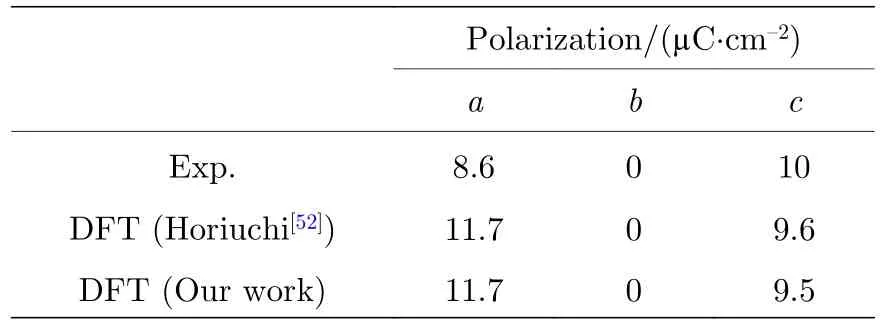
表1 铁电相CBDC 分子晶体的自发极化Table 1.Spontaneous polarization of ferroelectric phase of CBDC molecular crystal.
仔细观察CBDC 的铁电块体结构可以发现,其中的CBDC 分子自发排列于(102)晶面,若将该平面内的分子集合视为一单层结构,则CBDC分子晶体块体可视为这些单层结构的堆叠,如图5所示.为了探究CBDC 块体是否可被剥离为相应的二维结构,对块体沿着多个方向的剥离能进行了计算.本文使用公式ΔE=(Elayer-d—Ebulk)/(2S)对材料的剥离能进行量化计算,其中Ebulk为块体的能量,Elayer-d是相邻两单层之间相对于初始结构拉开距离d时体系的能量,S为剥离对应的横截面积.为了有更清晰的参照,计算了CBDC 块体沿着(100),(010),(010)和(102)晶面的剥离能,相应的结果如图6 所示.可以看出,沿着块体(100),(010),(001)和(102)平面的剥离能分别为0.37,0.37,0.33 和0.22 J/m2,显然沿着(102)晶面的剥离能显著小于其他晶面的对应值,仅约为石墨烯剥离能的2/3,意味着沿着CBDC 块体(102)晶面剥离出相应的二维结构是有可能的,本文将相应的二维结构命名为CBDC (102)单层.其实该结果通过分析分子晶体内部结构特性是显而易见的,相比于(100)或(001)晶面,沿着CBDC 块体的(102)晶面进行剥离并不会使晶体中的氢键链断裂,尽管沿着(010)晶面剥离块体亦未破坏氢键链,但该过程拉开了晶体中氧原子富集(负电荷富集)侧和氢原子富集(正电荷富集)侧,从而导致其剥离能相对较高.事实上,由图5 可明显看出,CBDC 块体的(102)晶面中的每个分子平面与(102)基面平行,呈现出明显的分层结构.

图5 CBDC 分子晶体块体沿(102)晶面的剥离示意图.其中左图中黄色平面代表(102)密勒晶面,右图为(102)单层的顶视图.灰色乳白色和红色小球分别指代碳原子、氢原子和氧原子.Fig.5.Exfoliation of CBDC bulk crystal along the (102) Millar plane.The yellow plane in left panel denotes the (102) plane.The right panel is the top view of the single-unit-thick (102) plane.The gray,white and red balls denote C,H and O atoms respectively.

图6 CBDC 块体沿着(100),(010),(010)和(102)晶面的剥离能,图中横坐标为相邻两单层之间相对于初始结构所拉开的距离Fig.6.Energy required to cleave the bulk of CBDC molecular crystal along (100),(010),(001) and (102) plane.The x axis denotes the interlayer distance of the two fractured sections compared with the original bulk crystal.
对于CBDC (102)分子单层,其中的氢键自发排列为氢键链并沿基矢a方向定向延伸,如图7(a)所示,推测该单层结构可能存在自发极化且自发极化沿着晶格的a方向.使用贝里相位方法计算单层CBDC (102)的铁电极化值,采用图7(b)中人为构造的拟顺电相作为参考相,通过线性插值的方式构建多个顺电相和铁电相间的中间结构,并以质子转移为序参量(λ),计算了相应的极化值,计算结果如图8 所示(λ=0 对应顺电相,λ=1对应铁电相).显然,图8 直观地得出了CBDC (102)分子单层具有自发极化,且其极化沿着基矢a方向,这与我们之前的推测是完全符合的.

图7 (a) 顺电相下的CBDC (102),绿色圆圈中质子处于相邻两个氧原子中间处;(b) 顺电相下的CBDC (102)中的CBDC 分子结构示意图,黄色平面为分子的对称面Fig.7.(a) Paraelectric phase of CBDC (102) monolayer crystal,the protons in green circles are located at the center of the adjacent oxygen atoms;(b) CBDC molecular in paraelectric phase,the yellow plane denotes symmetry plane of the molecular.
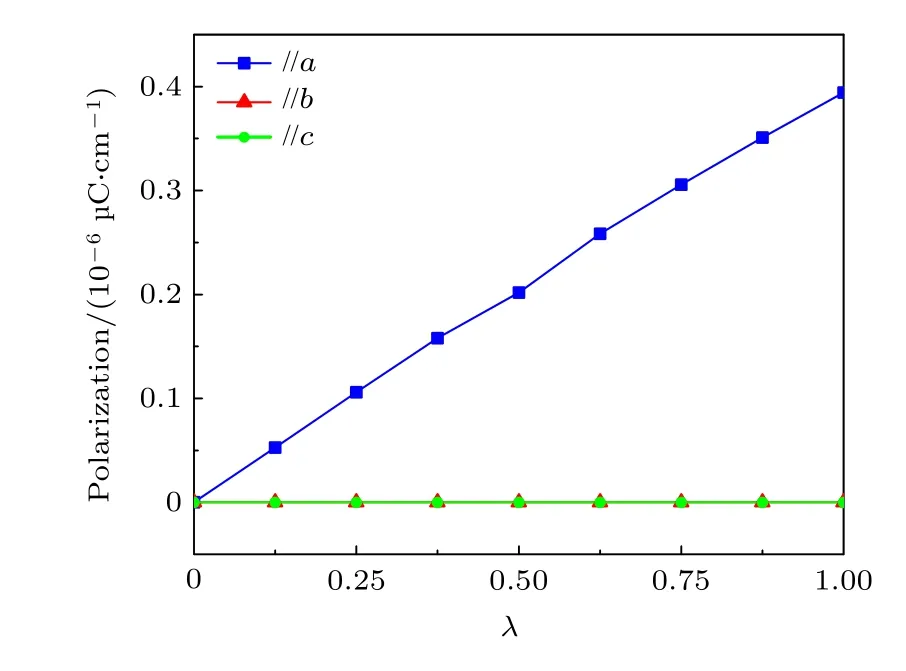
图8 CBDC (102)分子单层从顺电参考相(λ=0)转变至铁电相(λ=1)的过程中极化值的演变曲线Fig.8.Evolution curves of ferroelectric polarization for CB DC (102) monolayer crystal from the referenced paraelectric phase (λ=0) to ferroelectric phase (λ=1).
表2 列出了CBDC (102)单层的晶格参数和自发极化值.值得注意的是该单层分子晶体的极化值达到了0.39 × 10—6µC/cm,显著高于二维无机铁电材料中的Bi2O2S[30].

表2 CBDC (102)的晶格参数以及自发铁电极化值Table 2.Lattice parameters and spontaneous ferroelectric polarization of CBDC (102).
铁电材料的极化反转势垒(铁电极化发生反转P→ —P所需要的能量)是铁电材料的一项重要参数,势垒越高材料越不易受噪声电场的干扰,铁电-顺电转变温度(居里温度)往往也越高,因此铁电极化反转势垒在很大程度上反映了材料的铁电稳定性.本文在CBDC (102)分子单层的铁电相的极化反转(P→ —P)路径中线性地插入相应结构,并对其能量进行计算,从而得到相应的极化反转势垒.从图9 可以看出,极化反转路径能量与质子相对位置的变化曲线明显成对称的单峰特征,这表明本文选择的极化反转路径是对高维势能面上的最低能量路径的合理近似,其铁电极化反转势垒为0.76 eV/f.u..而双势阱曲线的存在再次印证了CBDC (102)单层分子晶体的铁电性.由于体系中氢键内质子的集体迁移是极化反转的直接原因,将势垒均分到参与迁移的质子上,约为0.095 eV/proton,该值远大于钙钛矿体系中的BaTiO3(极化反转势垒Ep≈ 0.009 eV/atom,Tc≈ 400 K)[16],意味着材料很有可能具备较高的居里温度,从而能抵抗高温下的热扰动.

图9 CBDC (102)铁电极化反转路径的能量变化曲线Fig.9.Energy profile along the polarity reverse path of the CBDC (102) monolayer crystal.
在实际的单层分子晶体制备中,往往会将分子晶体单层置于支撑衬底之上,单层分子晶体和衬底之间的晶格失配导致分子晶体单层受到外在的压缩或拉伸应力.为了探究CBDC (102)分子晶体单层的极化反转势垒对外加应力的响应,对CBDC(102)单层分子晶体施加平行于极化方向(即基矢a方向)的单轴应力,并计算在不同应力下极化反转势垒的变化.图10 和图11 分别为施加了1%—5%的单轴压缩应力(这里的一维应变定义为某一方向晶格长度变化量与原晶格长度的比例)和拉伸应力下材料的极化反转能量曲线.可以看出,伴随着单层CBDC (102)晶格的压缩,材料的极化反转势垒将降低,反之当其晶格被拉伸时势垒将增高.由于CBDC (102)中极化反转的实质是质子的定向迁移,故当极化方向上晶格的长度增加时质子迁移的路径将增加,从而导致势垒的增加,这显然是合乎规律的.从图10 和图11 可以看出,在5%的压缩应力之下,单层CBDC 的极化反转势垒为0.48 eV/f.u.,而在5%的拉伸应力下势垒升高至1.63 eV/f.u.,即0.20 eV/proton,因而材料对应力的响应十分灵敏,有望被应用到相应的铁电器件之中.
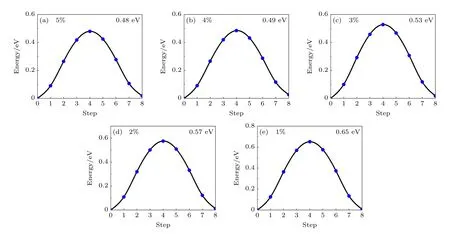
图10 平行于极化方向的单轴压缩应力下单层CBDC (102)的极化反转能量曲线Fig.10.Potential energy curve for the polarity reversal of the CBDC (102) monolayer crystal under uniaxial compressive strain range of 1%—5%.

图11 平行于极化方向的单轴拉伸应力下CBDC (102)单层分子晶体的极化反转能量曲线Fig.11.Potential energy curve for the polarity reversal of the CBDC (102) monolayer crystal under uniaxial tensile strain range of 1%—5%.
为了更直观地了解材料在单轴压缩或拉伸应力下极化反转势垒的变化趋势,将CBDC (102)单层分子晶体单胞的铁电极化反转势垒在不同应变(包括拉伸和压缩)下的数值变化进行整合,变化曲线如图12 所示.显然,材料中的氢键随着拉伸应力而延长,增加了铁电极化反转时质子的迁移路径长度,从而导致势垒不断提高.图12 中能量势垒在0.48—1.63 eV 之间,可知材料极化反转势垒对拉伸应变的响应相对更为敏感.

图12 二维CBDC (102)单层分子晶体在—5%—5%单轴应力下的极化反转势垒变化曲线Fig.12.Ferroelectric reversing barrier of two-dimensional CBDC (102) monolayer molecular crystal under uniaxial strain from —5% to 5%.
众所周知,铁电材料一般具有半导体特性,使得分散的正负电荷不会因自由移动而导致极化消失.这里也对单层CBDC (102)的能带结构进行了计算,结果如图13 所示,CBDC (102)分子单层为宽带隙半导体,其带隙值约为3.4 eV.

图13 单层CBDC (102)的能带结构(带隙值为3.4 eV)Fig.13.Band structure of CBDC (102) monolayer crystal.
由于CBDC (102)为单层有机分子晶体,其内部结合作用相对较弱,有限温度下的热涨落对结构稳定性的影响相比二维无机材料可能更为显著.因此,实验上在二维有机材料的生长与器件制备过程,往往会用化学惰性的石墨烯、六方硼氮或其他界面材料来支撑和稳定二维有机材料.本文提出的CBDC (102) 单层分子晶体在实际制备和应用的过程中也需要惰性衬底来支撑和稳定结构.因此,为了探究在室温下CBDC (102)单层分子晶体的铁电序能否稳定存在,将CBDC (102)单层分子晶体放置于石墨烯衬底上,在室温300 K 下进行了第一性原理的分子动力学模拟.在AIMD 计算中采用了2× 2的CBDC(102)超胞,为了减少晶格失配的影响,构建了的石墨烯衬底(CBDC超胞和石墨烯衬底的晶格失配在2%以下).对CBDC (102)单层分子晶体进行了5 ps 的分子动力学模拟,终态结构如图14 所示.可以看出,虽然CBDC (102) 单层分子晶体在垂直方向上结构发生了一定的起伏,但面内的定向氢键链的排列仍然规整有序,即在室温下该材料仍然保持了完整的面内铁电序,表明这种分子晶体单层的室温铁电性.
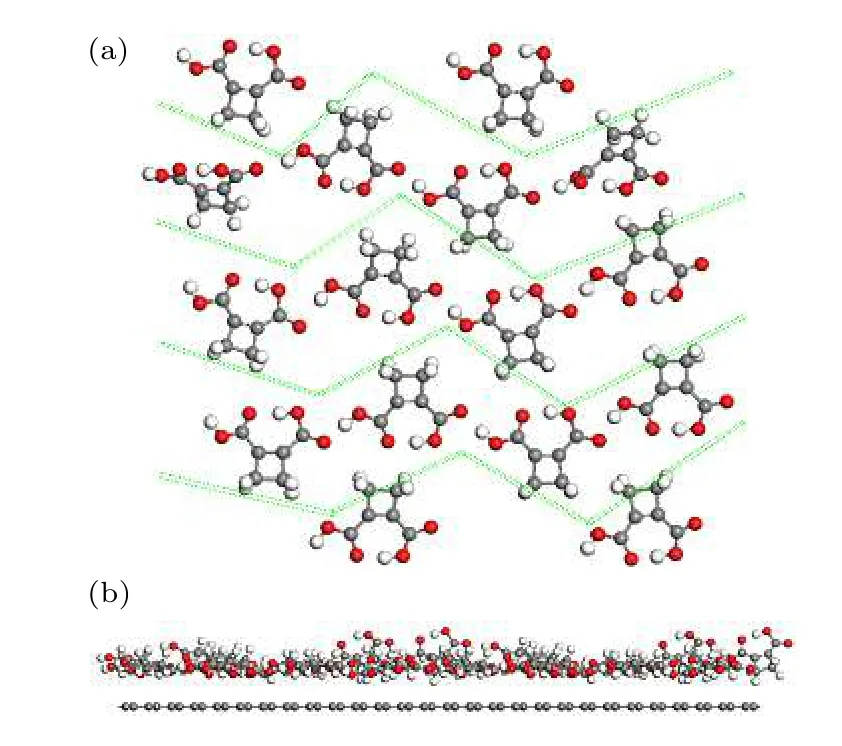
图14 石墨烯基底上的CBDC (102)单层分子晶体经过5 ps 从头算分子动力学模拟后的结构图(NPT 系综,温度300 K,时间步长1 fs) (a) 正视图;(b) 侧视图.图(a)中绿色曲线指示了多条有序排列的氢键链,为清晰起见,图(a)中的石墨烯衬底已被隐去Fig.14.(a) Top and (b) side views of snapshot of the twodimensional CBDC (102) monolayer supported on a graphene sheet,after 5 ps of AIMD simulation.The substrate in panel (a) is removed for clarity.
4 结论
本文基于第一性原理的理论计算设计了一种以CBDC 分子为结构单元的二维单层有机铁电分子晶体,即CBDC (102)分子晶体单层.通过计算其各向异性剥离能提出,该分子晶体单层有可能通过沿着块体的(102)晶面进行机械/化学剥离而获得.理论预测CBDC (102)分子晶体单层的自发铁电极化值约0.39 × 10—6µC/cm,可与部分二维无机铁电材料相比拟.计算表明CBDC (102)分子晶体单层的铁电极化反转势垒达到了0.76 eV/f.u.,显著高于钙钛矿体系中BaTiO3,预示了该分子晶体单层有望展现出良好的鲁棒性和较高的铁电转变温度.模拟发现CBDC (102)分子晶体单层的极化反转势垒对外加单轴应力的响应较为敏感,有望通过界面应力工程对CBDC (102)分子晶体单层的极化反转势垒进行调控.从头算分子动力学模拟表明CBDC (102)分子晶体单层具有良好的热稳定性,其铁电序在室温下可以很好地保持.CBDC(102)单层有机铁电分子晶体的高面内自发极化和易被界面调控的极化反转势垒,以及良好的热稳定性使其可能被应用于轻质无金属及柔性铁电器件.
