核壳纳米材料制备及其在CO/CO2热催化加氢中的应用
2022-03-24金科王晨光马隆龙张琦
金科,王晨光,马隆龙,张琦
(1中国科学院广州能源研究所,中国 科学院可再生能源重点实验室,广东 广州 510640; 2东南大学能源与环境学院,能源热转换及其过程测控教育部重点实验室,江苏 南京 210096; 3中国科学院大学,北京 100871)
引 言
能源危机和环境问题使人们迫切希望找到可替代传统化石能源的燃料。将浓度逐年攀升的温室气体CO2进行加氢和由生物质等可再生资源转化得到的合成气(H2+CO)进行费托合成(FTS)反应,可制备CH4、低碳烯烃、醇、芳烃、汽油和煤油等多种化学品[1],该方案既可有效缓解温室效应,又可对能源危机提供解决思路。CO/CO2热催化加氢反应的关键在于C—O 键活化与C-C 偶联的精确控制。CO分子相对活泼,极容易被Fe、Co 和Ru等费托催化剂的表面吸附、活化并发生C-C 偶联,其产物分布服从Anderson-Schulz-Flory(ASF)分布[2],费托催化剂与分子筛耦合可提高目标产物选择性(费托路线)。同时,CO 也可被Zn、Cr和Zr等金属氧化物的表面吸附、活化并生成甲醇等含氧中间体,然后在分子筛中进行C-C 偶联合成高值化学品(甲醇路线),其产物分布不受ASF 模型限制[3]。CO2的活化则较为困难,一般需要输入高能量和/或高密度的还原剂才能打破C—O 键的壁垒。CO2也可通过甲醇路线和改进的费托路线转化为高值化学品[4]。目前,CO/CO2热催化加氢反应催化剂的结构多为负载型、体相型和物理混合型。可进行CO/CO2活化加氢甚至中间体C-C 偶联的负载型和体相型金属催化剂包括含氧中间体合成催化剂和费托催化剂。可实现接力催化的负载型和物理混合型双功能催化剂主要包括两种组合:一是含氧中间体合成催化剂与分子筛的耦合;二是费托催化剂和分子筛的耦合。然而,常规负载型和体相型金属催化剂在CO/CO2热催化加氢反应过程中往往存在活性不够高、稳定性差和目标产物选择性低等问题。例如,在CO2加氢制甲醇过程中,由于常规的铜基催化剂上Cu的结合能较弱,在工作条件下容易重构、团聚甚至烧结[5-6]。在CO2加氢制取烃类化合物过程中,负载型Fe-K/Al2O3催化剂的Fe5C2活性相容易转变为Fe3C而失活[7]。常规负载型和物理混合型双功能催化剂虽能实现接力催化,但其提供的是一个开放和不受限制的反应环境,两类反应是随机和独立发生的。在含氧中间体合成催化剂或费托催化剂上产生的中间体向分子筛的扩散效率仍处于一个相对较低的水平,这使目标产物的选择性和产率被极大限制[8-9]。
核壳催化剂的设计为克服上述问题提供一种切实可行的方法。核壳催化剂是由中心的核体和外部的壳层通过化学键或其他相互作用包覆形成的有序组装结构的复合材料。它可将多种材料整合到一个功能体系中,表现出良好的物理化学性质(如稳定性、无毒性、分散性、多功能等)[10]。更为重要的是,核壳纳米材料内部各组分之间的活性界面可能会产生优异的协同作用和新的性能,这使其在催化、吸附、储能与转化、药物传递和光学等方面具有广阔的应用前景[11-14]。相比于常规的CO/CO2加氢催化剂,核壳催化剂的壳层包覆可对核体粒子表面进行修饰,如改变其表面电荷、官能团和反应特性等,从而提高核体的稳定性与分散性。核壳催化剂还可形成封闭的内部微环境以富集反应物,提高反应速率和催化活性。部分核壳催化剂甚至还能实现接力催化,并提高体系内的能量利用率。
本文主要介绍了核壳纳米材料的常用制备方法,不同类型壳层包覆的核壳催化剂在CO/CO2热催化加氢中的应用进展,并对该领域的未来发展进行了展望。
1 核壳纳米材料的合成策略
核壳纳米材料的制备方法在一定程度上决定其结构和性能,采用不同方法制备的核壳纳米材料在微观结构、形貌、孔体积、比表面积和化学性能等方面存在较大的差异。目前,制备核壳纳米材料的方法主要有溶胶-凝胶法、水/溶剂热法、微乳液法、化学气相沉积法、层层组装法、热分解法和机械球磨法等。
1.1 溶胶-凝胶法
溶胶-凝胶法就是将含高化学活性组分的化合物经过溶液、溶胶、凝胶和凝胶固化,再经热处理而成氧化物或其他化合物固体的方法。以钛酸乙酯为前体、羟基丙基纤维素为表面活性剂和空间稳定剂,采用溶胶-凝胶法可实现非晶态TiO2对单分散SiO2球的均匀包覆,TiO2壳层厚度可通过调整核的大小(硅球)、反应时间、水和表面活性剂的浓度来控制,非晶态TiO2壳层经热处理可转化为具有高机械强度的金红石,合成的SiO2@TiO2核壳材料具有三维有序结构和高折射率[15]。以十六烷基三甲基溴化铵(CTAB)为模板,采用溶胶-凝胶法可在Fe3O4@nSiO2微球上包覆一层CTAB/SiO2复合材料,然后通过丙酮抽提将CTAB模板以温和的方式去除,形成介孔无定形SiO2壳层,所制备的Fe3O4@nSiO2@mSiO2微球具有超顺磁性、均一的介孔孔道、高比表面积和大孔容等优点(图1)[16]。此外,溶胶-凝胶法还被广泛用于合成其他功能介孔纳米球,如聚合物和碳等[17-20]。该方法具有制备温度低、化学均匀性好、过程易于控制和产品纯度高等优点。但也存在原料价格昂贵、部分原料有毒害、周期长等不足。
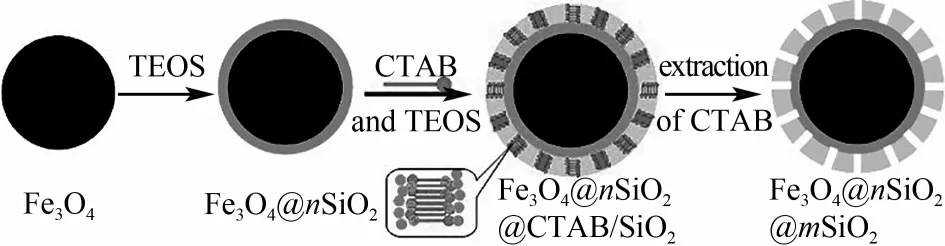
图1 Fe3O4@nSiO2@mSiO2微球形成原理图[16]Fig.1 Formation principle diagram of Fe3O4@nSiO2@mSiO2 microspheres[16]
1.2 水/溶剂热法
水/溶剂热法合成核壳纳米材料的过程是在封闭的高温、高压下以有限的体积进行的,这种条件能够实现常规条件下无法进行的反应,通过改变工艺条件可合成特定晶体结构、组成、形貌以及颗粒尺寸的产物。以单分散非球形赤铁矿α-Fe2O3为核和TiF4为前体,通过水热法可合成α-Fe2O3@TiO2核壳纳米颗粒,水热过程中PVP 有助于锐钛矿型TiO2纳米粒子均匀、稳定地沉积在大型纳米核上[21]。以葡萄糖为碳源,采用水热法可合成粒径从几百纳米到2 μm 范围的单分散Ag@C 和Au@C 核壳纳米粒子,其粒径可通过改变反应时间、温度和葡萄糖的起始浓度来调控[22]。水热法也可用于FexOy@C 的合成,硝酸铁前体的加入可大幅降低葡萄糖的水热炭化温度(<160℃),得到具有特殊结构的布丁型FexOy@C 微球(图2)[23],其无定形碳壳既可促进核体粒子的还原又可保持其结构稳定。

图2 水热法合成FexOy@C微球的流程示意图[23]Fig.2 Schematic diagram of FexOy@C microspheres prepared by hydrothermal method[23]
与水热法类似,溶剂热法采用有机溶剂或非水溶媒代替水,可制备出在水溶液中无法合成、易氧化、易水解和对水敏感的材料。例如,在氧化铁纳米粒子的合成过程中,如果采用水热法产物为纺锤形Fe2O3纳米粒子[24]。当把溶剂换成乙二醇时,产物则为球形Fe3O4纳米粒子[25]。在含葡萄糖、Fe3+、乙二醇和尿素的溶剂热反应体系中,尿素分解产生OH-与Fe3+形成Fe(OH)3;同时,葡萄糖发生脱水聚合,聚合产物通过氢键等作用力吸附在Fe(OH)3表面,并阻碍晶体的团聚,形成高度均一分散的胶体结构;随着部分Fe(OH)3被乙二醇还原为Fe(OH)2,两晶核相互结合脱水形成Fe3O4晶粒,吸附在表面的葡萄糖脱水产物进一步聚合炭化,形成分散的胶体粒子;在持续加热条件下,最终形成无定形碳质外壳包裹Fe3O4的纳米粒子(图3)[26]。

图3 溶剂热法制备Fe3O4@C核壳纳米粒子的流程示意图[26]Fig.3 Schematic diagram of Fe3O4@C core-shell nanoparticles prepared by solvothermal method[26]
采用水/溶剂热法合成核壳纳米材料操作简单,利于制备对空气敏感的物质,产物粒径小、分布均匀、团聚程度较轻、纯度较高。相比于其他方法,水/溶剂热法不需要高温煅烧晶化,避免了杂质的引入和结构缺陷,但对设备的要求较高。
1.3 微乳液法
微乳液法是两种互不相溶的溶剂在表面活性剂的作用下形成乳液,在微泡中经成核、聚结、团聚、热处理后得到纳米粒子。通过改变微乳液体系中水/表面活性剂的摩尔比可调节胶束的大小和形状,进而调控核壳纳米粒子的粒径。室温下,采用微乳液法制备高单分散Co@Ag 核壳纳米粒子的流程如图4所示[27],首先将含有Co(ClO4)2和还原剂的两种胶体溶液等体积混合,胶束之间的快速物质交换使得Co 核迅速生长。其次在含钴核的微乳液中分别倒入等体积的含AgClO4和还原剂的两种胶束溶液。完全还原后,向溶液中加入丙酮,使其发生相分离。最后用丙酮洗去表面活性剂即可得到Co@Ag 核壳纳米粒子,其粒径可通过改变微乳液体系中水/表面活性剂的摩尔比来调控。采用微乳液法制备Pt@SiO2纳米颗粒时,其SiO2壳层厚度可通过TEOS 的用量来调控。相比于负载型的Pt/SiO2催化剂,具有膜作用的富微孔无定形SiO2壳层可提高Pt表面O2/CO 值,使CO 的氧化速率提高了一个数量级[28]。该方法具有操作简单、能耗低、产物粒径可控、粒子间不易聚集等优点。但制备过程中,表面活性剂和稳定剂等化学物质可能会集成在外壳层中,从而降低核壳材料的热稳定性。
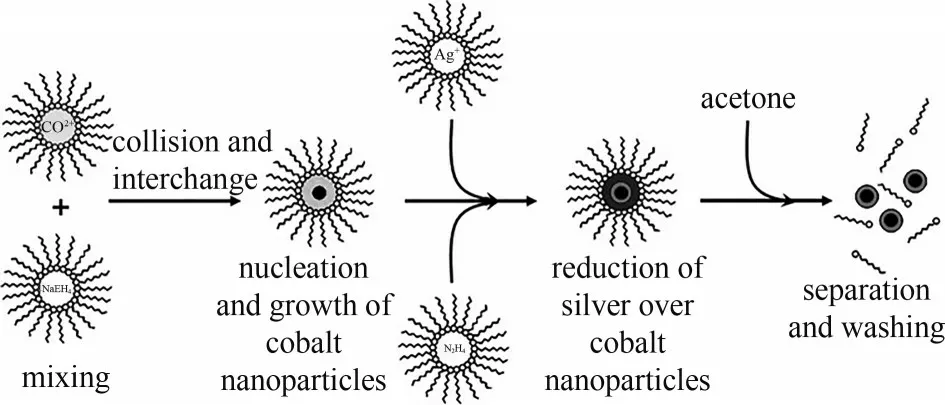
图4 微乳液法制备Co@Ag核壳纳米粒子的流程示意图[27]Fig.4 Schematic diagram of Co@Ag core-shell nanoparticles prepared by microemulsion method[27]
1.4 化学气相沉积法
化学气相沉积法是利用气态物质在固体表面进行化学反应(热分解或者化学合成),生成固体沉积物的过程。采用化学气相沉积法可制备石墨烯包覆纳米Al2O3核壳材料[29],首先纳米Al2O3表面在混合气氛(C2H2、H2和Ar)中被还原形成薄铝层,然后C2H2在铝层表面分解生成石墨烯壳层,其层数随沉积反应时间延长而增加。在制备无任何污染和结晶性良好的Cu@C 纳米线时(图5)[30],石墨碳壳层的厚度也可通过沉积反应时间来调控,该Cu@C 纳米线比裸铜纳米线具有更好的抗氧化性能。该方法具有淀积温度低、薄膜成分和厚度易控、均匀性和重复性好等优点。但也存在沉积率不高,沉积反应源和余气易燃、易爆,对设备耐腐蚀性要求较高等不足。

图5 化学气相沉积法制备Cu@C核壳纳米材料的流程示意图[30]Fig.5 Schematic diagram of Cu@C core-shell nanomaterials prepared by chemical vapor deposition[30]
1.5 层层组装法
层层组装法是借助各层分子间的静电吸引力、氢键和配位键等弱相互作用使层间分子自发地缔合形成结构完整、性能稳定、具有某种特定功能的分子聚集体或超分子结构的过程。多次层层交替沉积可实现复合物涂层的均匀包覆(图6),复合材料的壳层厚度可通过改变聚合电解质的沉淀次数或浓度来调控[31]。以二(2-羟基丙酸)二氢氧化铵合钛作为前体,氯化聚二烯丙基二甲基铵作为聚电解质,采用层层组装法可合成锐钛矿型TiO2壳层包覆Au纳米粒子的核壳纳米材料,并通过改变沉积次数有效控制膜层厚度[32]。通过层层组装法可实现将预先合成的Au 胶体和聚乙烯亚胺电解质沉积在Fe3O4@SiO2磁性微球上,所制备的球形核壳双功能材料在有机和无机还原反应中均表现出良好的催化性能[33]。该方法制备简单、产物壳层厚度可控、应用广泛。但一般耗时较长,且在反复沉积过程中容易导致沉积液的交叉污染。

图6 层层自组装法的流程示意图[31]Fig.6 The process diagram of layer-by-layer self-assembly[31]
1.6 热分解法
热分解法是将易分解的金属配合物在高温下分解形成具有核壳结构复合材料的过程。采用热分解法可实现不同形貌Cu2O@Ag 核壳多面体纳米颗粒的可控制备,纳米颗粒的壳层性质和表面银纳米颗粒的粒径可通过改变热分解条件来调控[34]。具有大的比表面积、孔容和孔径分布明确等特点的MOFs 是制备核壳纳米材料的理想前体。热解MOFs 前体制备的多孔复合材料可继承其部分甚至全部的优良性能[35]。通过对钴基普鲁士蓝纳米立方体的自组装和可控热分解,可制备均匀分布的CoFe@C 核壳纳米粒子(图7),其颗粒尺寸还可通过改变MOFs 前体的晶粒大小来调控,石墨碳层的包覆使该核壳材料具有良好的导电性和结构稳定性[36]。热分解法操作十分简单、产物结晶度好、粒径分布较窄、适合大批量生产。但受限于前体类型的选择,合成成本相对较高,适用的体系有限。

图7 热分解法制备CoFe@C核壳纳米粒子的流程示意图[36]Fig.7 Schematic diagram of CoFe@C core-shell nanoparticles prepared by thermal decomposition method[36]
1.7 机械球磨法
机械球磨制备核壳纳米材料是一种过程简单且干净的方法。将Pd 金属纳米颗粒与CeO2多晶粉末在干燥条件下球磨混合,可合成Pd@CeO2核壳催化剂(图8),其Pd-O-Ce 活性界面使CH4氧化反应的光照温度降低了50℃以上,并提高了反应速率[37]。在Al2O3@Co 核壳催化剂的制备过程中,改变球磨时间可以调节复合材料的粒径和壳层中钴相(hcp/fcc)的比率[38]。该过程避免了有机溶剂和其他添加剂的使用,所制备的核壳材料具有较高的纯度。但也会因壳层颗粒间团聚造成材料利用率有限等不足。

图8 机械球磨法制备Pd@CeO2核壳催化剂的流程示意图[37]Fig.8 Schematic diagram of Pd@CeO2 core-shell catalyst prepared by mechanical ball milling[37]
1.8 其他方法
除了上述方法外,还有超声化学法[39]、非均相成核法[40]、电弧放电法、离子交换法、化学镀法[41]和辐照合成法[42]等。需要指出的是,多种制备方法的结合可有效调控制备过程,获得性能更好、应用更有针对性的核壳结构材料。
2 核壳催化剂在CO/CO2热催化加氢中的应用
在CO/CO2热催化加氢反应中,常规负载型、体相型和物理混合型催化剂往往存在活性不够高、稳定性差和目标产物选择性低等问题。而核壳催化剂的设计为克服上述问题提供一种切实可行的方法。目前,CO/CO2热催化加氢反应中核壳催化剂的壳层材料主要包括金属单质壳、金属氧化物壳、SiO2壳、碳壳和分子筛壳等。
2.1 金属单质壳
Pd、Co 和Ru 等金属单质是CO/CO2热催化加氢反应的活性相,将其作为催化剂壳层可提高金属单质的利用率和核体催化剂的还原度,同时壳层金属单质可与核体催化剂协同实现接力催化,从而达到调控催化活性和目标产物选择性的目的。
Ag@Pd-ZnO 核壳催化剂中富电子的Pd 壳层和PdZn 相使得每克Pd 对CO2加氢制甲醇的催化效率比普通的Pd-ZnO 催化剂高3.32 倍,同时核壳结构可减少贵金属Pd 的用量[43]。Co 颗粒包覆Fe5C2的核壳催化剂可实现CO 加氢制C+5的接力催化,Co 负责CO 的解离,Fe5C2负责220℃下的碳链增长,两者协同可提高FTS 反应的性能。在0.6%(质量分数)的Co(Fe/Co=12)加入时,Fe5C2/Co核壳催化剂的低温活性是纯Fe5C2催化剂的4倍[44]。CeO2-Pt@mSiO2-Co核壳催化剂可实现对CO2加氢制C2~C4烃类的接力催化(图9),CO2首先在Pt/CeO2界面上发生逆水煤气变换(RWGS)反应转化为CO,生成的CO 在相邻的Co/mSiO2界面上发生FTS 反应生成C2~C4烃类产物,该催化剂具有比CeO2-Pt@mSiO2、CeO2@mSiO2-Co和物理混合物催化剂更高的C2~C4烃类选择性和稳定性(40 h),同时催化剂表面较低的H2/CO2能够减弱Co上甲烷化反应的发生,促进接力催化反应的进行[45]。在CO2加氢制长碳链烃反应中,Co@Ru/γ-Al2O3催化剂的特殊核壳结构和Ru 包覆量的增加可降低CoO 物种的还原温度,在150~400℃的H2程序升温还原(H2-TPR)中,还原度由68%显著提高到98.7%。Co@Ru 中核壳间原子的强电子相互作用使催化剂的FTS性能得到大幅度提高。同时随着壳层厚度的增加,CO转化率和重碳烃选择性显著提高[46]。

图9 接力催化剂的结构图及其催化性能[45]Fig.9 Structure diagram of relay catalyst and its catalytic performance[45]
综上所述,金属单质壳包覆的核壳催化剂在CO/CO2热催化加氢反应中具有十分重要的作用,后期应重点关注于开发新的合成工艺来实现金属单质壳层的完全包覆,或者通过在单质壳层外再构建一层完全包覆的壳层,来避免金属单质壳层颗粒在高温反应中发生团聚和迁移,同时提高核体初级反应产物向壳层活性位点的迁移效率。
2.2 金属氧化物壳
金属氧化物作为催化剂壳层可改变核体催化剂的电子效应和表面碱度,调节催化剂对CO、CO2和H2的吸附量及吸附强度,从而实现调控催化活性和目标产物选择性的目的。同时,部分金属氧化物包覆可防止核体粒子发生团聚失活。
碱金属氧化物是良好的电子型助剂,Na2O 包覆铁催化剂可提高其表面碱度,促进Fe5C2活性相的生成,并提高Fe5C2活性相在CO2热催化加氢反应中的稳定性[47]。
MnO2既是结构型助剂又是电子型助剂,能够提高催化剂的分散度,抑制催化剂因积炭而失活。在CO/CO2热催化加氢反应中,助剂Mn 可促进单质铁渗碳形成Fe5C2活性相,其对Fe 基催化剂提供电子可减少表面铁物种的电子空穴,从而抑制加氢反应活性[48]。Fe2O3@MnO2催化剂的核壳结构可有效避免Fe-Mn 尖晶石氧化物的形成,并最大限度发挥Mn 提供电子的能力。在最优条件下,该催化剂上CO 转化率为86%,产物中C+5烃类选择性达到66.6%[49]。在FTS制线形α-烯烃过程中,Fe3O4@MnO2催化剂中壳层MnO2向核体Fe3O4的电子转移有利于CO分子在Fe3O4上的解离吸附和H原子在MnO2上溢出,从而增强催化剂上的C-C偶联概率,并减弱其加氢活性,这使其对烯烃(79.6%)和线形α-烯烃(59.1%)具有较高的选择性,同时MnO2壳层使催化剂在100 h内具有稳定且优越的催化活性(图10)[50]。MnO2包覆Fe3O4微球还可大幅度提高催化剂的比表面积,为CO2吸附和C—O 键活化提供更多的位点,这使10Mn-Fe3O4催化剂对低碳烯烃的选择性和产率分别达到46.2%和18.7%,而副产物CO选择性低至9.4%[51]。

图10 Fe3O4@Mn-50催化剂上的烃类分布及其稳定性[50]Fig.10 Hydrocarbon distribution and stability over Fe3O4@Mn-50 catalyst[50]
具有氧化还原特性和高氧迁移率的CeO2被广泛用于CO2和CH4干重整。在苛刻的干重整反应条件下,Ni@CeO2催化剂的CeO2壳层可抑制纳米镍的烧结和生长,提供更多的晶格氧和空位,并抑制焦炭沉积,这使得Ni@CeO2核壳催化剂在CO2和CH4干重整反应中表现出比Ni/CeO2等负载型催化剂更好的催化活性和稳定性[52]。
Cu/ZnO 催化剂的Cu-ZnO 接触界面对CO2加氢制甲醇具有较高的活性。采用ZnO 包覆的核壳结构催化剂可实现Cu-ZnO 接触界面的最大化,加速Zn 向Cu 扩散,在异质接界处产生大量活性位点。在催化剂还原过程中,Cu@ZnOx核壳催化剂的Zn 会迁移到Cu 核上形成缺氧ZnOx或CuxZn1-xOy层,缺氧层通过吸附CO2和Cu 纳米粒子上溢出的氢作用为甲醇的形成提供活性位点。缺氧层对Cu 的完全包覆阻碍了RWGS 反应和甲醇分解过程中CO 的生成,这些使Cu@ZnOx核壳催化剂对甲醇的选择性达到100%[53]。工业用的Cu/ZnO/Al2O3催化剂在反应过程中还会发生Cu0相和ZnO 相的分离,进而导致Cu0的团聚和Cu0-ZnO活性界面的收缩,降低催化活性。而介孔SiO2和Al2O3壳包裹Cu/ZnO/Al2O3催化剂(CZAS@SA)可有效抑制Cu0相和ZnO 相的分离,使催化剂在反应过程中保持较高的甲醇选择性。且研究发现CZAS@SA 催化剂还表现出比Cu/ZnO/Al2O3催化剂更高的二甲醚(DME)、甲醇生成本征活性和低得多的CO生成本征活性,使产物中二甲醚和甲醇的总选择性由9.1%(摩尔分数)提高到63.3%[54]。
具有良好热化学稳定性和优异电子性能的TiO2也被用来构建纳米核壳催化剂。Co-Z@TiO2催化剂的多孔TiO2壳既可加强扩散限制,使FTS中间产物进一步聚合为高碳烃,还可使催化剂在110 h的FTS反应中保持较好的催化性能和钴的粒径分布。在最优条件下,Co-Z@TiO2(N2+O2)催化剂具有较高的钴时空产率[14.84 μmol CO/(g Fe·s)] 和C+5选择性(81.5%)[55]。在FTS 反应中,钛纳米管包覆有利于核体铁物种的还原性,增强短链α-烯烃在钛纳米管内部狭窄空间内再吸附,这使Fe2O3@TiO2催化剂对和烃类的选择性远高于钛纳米管外负载铁纳米粒子的催化剂[56]。TiO2包覆还可使Cu@TiO2核壳催化剂的催化活性比单独的Cu纳米粒子高出20倍,且经过高温RWGS反应后其核壳构型不变(图11)[57]。
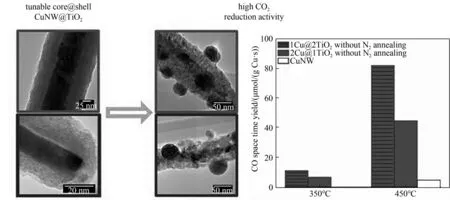
图11 Cu@TiO2催化剂在反应前后的结构变化及其在不同温度下的催化活性[57]Fig.11 Structure change of Cu@TiO2 catalyst before and after reaction and its catalytic activity at different temperatures[57]
2.3 SiO2壳
具有良好的机械性、热稳定性和化学惰性的SiO2是应用最广泛的壳层材料之一。采用SiO2包覆金属或金属氧化物既能增强活性金属的分散度和核壳结构的机械强度,又可抑制活性组分烧结。在FTS反应中,SiO2壳层保护的Fe3O4@SiO2催化剂具有更高的反应活性和稳定性,经100 h 反应后其核壳结构基本不变[58]。具有双壳层结构的FeSiMn 催化剂中SiO2壳层不仅为MnO2纳米颗粒提供了锚定位点,而且避免了铁锰尖晶石的形成(图12),这使其在FTS 反应中具有较高的催化活性[3.41×10-5mol CO/(g Fe·s)]和C+5产率[20.8×10-5mol HC/(g Fe·s)][59]。FeMn@SiO2催化剂中SiO2壳层的空间限域和氢富集的平衡作用可实现对C+5烃类选择性的有效调节,在最优条件下,FeMn@SiO2-0.25 催化剂上CO 转化率为67.2%,产物中C+5和烯烃的选择性分别达到71.8%和72.5%[60]。Co3O4@SiO2催化剂的SiO2壳层可有效抑制FTS 反应过程中钴纳米晶的聚集,通过控制钴的粒径即可实现对产物及其选择性的稳定调控。相比于负载型催化剂,壳层的限域效应可增加反应中间体和活性位点间的接触时间,进而促进链增长[61]。同时,纳米多孔SiO2壳层的择型性可缩短碳链分布[62]。CuIn@SiO2催化剂的SiO2壳层可使其在CO2加氢制甲醇反应中具有极高的稳定性和优异的催化性能,在最优条件下,CH3OH选择性和时空收率分别达到78.1%和13.7 mmol CH3OH/(h·g cat)[63]。介孔SiO2壳包覆Cu 和Cu/ZnO 纳米颗粒可抑制Cu纳米颗粒的团聚,使核壳Cu/ZnO@m-SiO2催化剂实现了CO2加氢制甲醇的高选择性(66.6%)和稳定性(168 h)转化(图13)[64]。

图12 FeSiMn双壳层催化剂形成的示意图及其催化性能[59]Fig.12 Schematic illustration of the formation of FeSiMn double-shell catalyst and its catalytic performance[59]

图13 Cu/m-SiO2(三角形)、Cu@m-SiO2(矩形)和Cu/ZnO@m-SiO2(圆形)催化剂上CO2转化率随反应时间的变化[64]Fig.13 The CO2 conversion with reaction time over Cu/m-SiO2(triangle),Cu@m-SiO2(rectangle)and Cu/ZnO@m-SiO2(circle)catalysts[64]
同时,SiO2壳层表面非常容易功能化。经过疏水处理的SiO2壳层在CO/CO2加氢反应中可及时移除RWGS 和FTS 反应过程中产生的水,使反应平衡向正反应方向移动。FeK/Al2O3@SiO2催化剂的疏水SiO2壳层可抑制反应过程中副产物水的竞争吸附,而且疏水SiO2的包覆量会影响催化剂活性和C+2烃类的选择性[65],当包覆量从0 增加到9%(质量分数)时,CO2的转化率由49%升高至63%,C+2烃类的选择性由63%上升至74%。甲基改性的Fe2O3@SiO2核壳催化剂对CO 加氢反应具有较高的活性和稳定性,疏水SiO2表面能抑制水的重吸水,降低水气变换反应(WGS)活性,从而抑制CO2的生成,该催化剂的SiO2壳层是连接Fe核和疏水甲基的桥梁[66]。在上述研究的基础上,Xu 等[67]进一步设计了一种具有良好疏水性的FeMn@Si 核壳催化剂(图14)。在FTS 反应中,疏水SiO2壳层通过缩短水在催化剂表面的滞留时间抑制了与水有关的副反应,从而既抑制了CO2副产物的生成又保护了碳化铁核体(Fe5C2)不被氧化。此外,锰原子向铁原子的电子转移促进了烯烃的生成,抑制了CH4的生成。在最佳条件下,该催化剂上副产物CO2和CH4的总选择性被抑制在22.5%以下,同时烯烃产率达到36.6%。

图14 Fe@Si、Fe@Si-c催化剂的结构及疏水性改性抑制CO2生成的机理[67]Fig.14 Structural of the Fe@Si,Fe@Si-c catalysts and plausible mechanism of hydrophobic modification inhibiting the CO2 formation[67]
采用SiO2包覆构建的核壳催化剂在CO/CO2加氢反应中具有很多优点,当反应过程中核体体积会发生剧烈膨胀时,可考虑在壳层与核体之间构建一定的缓冲空间,避免核体将SiO2壳撑破而失去保护作用。
2.4 碳壳
具有较好的抗腐蚀性、导电性、导热性、韧性和化学稳定性等优点的碳材料是一类很理想的壳层材料,它不仅可以抑制核体粒子继续长大和团聚,还可将碳层的优异性能赋予被包裹的粒子。在FTS反应中,2K-Fe3C@C 催化剂上类石墨烯壳层的富电子特性使得氢难以对不饱和中间体进行加氢,从而具有比普通铁基催化剂更高的低碳烯烃选择性(41.9%)。同时类石墨烯碳层可抑制铁纳米颗粒在苛刻条件下的迁移和聚集,使催化剂的形貌和性能在100 h 的FTS 过程中保持稳定[68]。在常规FTS 中,铁基催化剂的FexC 活性相容易发生相转变失活[图15(a)][69],而石墨烯壳层包覆可抑制ε-Fe2C在高温下的相变和积炭[图15(b)],这使得ε-Fe2C@C 核壳催化剂在300℃、10 bar(1 bar=105Pa)、H2/CO=1和16 L/(h·g)条件下的FTY 值高达1258 μmol CO/(g Fe·s),比传统的碳负载Fe 催化剂高出一个数量级[图15(c)],且在400 h 内无失活现象发生[图15(d)]。Co@C 催化剂的独特核壳结构可提供较高的吸附空间,石墨碳层缺陷有利于H2的解离吸附,增强金属Co 与CO 分子间的电子导电性。同时石墨碳壳还可抑制钴纳米粒子在活化和反应过程中发生聚集,这些使得该催化剂具有较高的稳定性和C+5选择性(56.8%),高于传统活性炭负载钴催化剂(Co/AC)的效果(46.2%)[70]。

图15 铁基催化剂费托合成的原理模型及不同铁基催化剂的催化性能[69]Fig.15 Schematic models of iron-based catalysts for Fischer-Tropsch synthesis and catalytic performance of different iron catalysts[69]
碳纳米管(CNTs)有明确的中空内部,管内电子云密度小于管外,表现出不同寻常的机械性、热稳定性及导电性。将其他物质引入空腔也可构建兼具其独特性能的核壳催化剂。Bao 等[71-72]发现CNTs限域的Fe2O3粒子(Fe-in-CNTs)还原为金属Fe 的温度比分散于管外壁的Fe2O3粒子(Fe-out-CNTs)低200℃,且随着管内径的减小[图16(a)],其还原温度逐渐降低,这种特性使CNTs 限域的铁纳米粒子在FTS反应条件下更容易转化为Fe5C2活性相[图16(b)],因此Fe-in-CNTs 催化剂的C+5烃收率是Fe-out-CNTs催化剂的2 倍,是Fe/AC 催化剂的6 倍以上(表1)[73]。CNTs 的限域效应还能提高FeN-in-CNTs 催化剂中N 在FTS 反应过程中的保留能力,从而提高催化剂的抗氧化能力,这使得FeN-in-CNTs 催化剂的活性比还原Fe-in-CNTs 催化剂和负载型FeN/SiO2催化剂高5~7 倍,而且位于CNTs 内的FeN 纳米粒子催化活性是管外负载FeN 纳米粒子的1.4 倍[74]。豆荚状CNTs 包覆铁纳米颗粒的核壳催化剂具有较高的低碳烯烃选择性(45%)和稳定性(120 h),而负载在豆荚状CNTs外的Fe基催化剂虽然也能获得42%的低碳烯烃选择性,但极易出现因Fe纳米颗粒的团聚和积炭而失活的现象[75]。CNTs 的限域效应还可使Rh粒子的尺寸和反应活性在CO 加氢制乙醇反应过程中保持稳定,缺电子的CNTs 内壁对Rh 粒子的修饰作用有利于倾斜式吸附CO 物种的形成,促进CO 的解离和C2含氧化合物的生成,这使得Rh-in-CNTs催化剂上乙醇的总生成速率[30.0 mol/(mol Rh·h)]比在Rh-out-CNTs 催化剂上的总生成速率高出一个数量级以上[76]。
A组手术时间、术中出血量、肠功能恢复时间均明显低于B组,两组比较差异有统计学意义(P<0.05)。见表1。

图16 Fe2O3/CNTs复合材料TPR过程中CO的演变及Fe-CNTs催化剂在接近FTS条件下的晶相演变[72-73]Fig.16 CO evolution during TPR of the Fe2O3/CNTs composites and crystalline phase evolution of Fe-CNTs catalyst near FTS condition[72-73]

表1 FTS活性和产物选择性的比较[73]Table 1 Comparison of the FTS activity and product selectivities[73]
在FTS 反应中,FeMn@C 核壳催化剂的壳层碳与中心铁锰的结合能够促进碳化铁活性相的形成,同时碳壳的限域效应能促进FTS产生的低碳烯烃中间体发生进一步聚合,这些使得该核壳催化剂具有比传统的FeMn/SiO2催化剂更高的催化活性、稳定性和C+5烃类(尤其是C5~C12烃类)选择性[77]。MOFs 衍生的碳基核壳纳米材料在FTS过程中也显示出无与伦比的性能。热解富氮的ZIF-67 和无氮的Co-MOF-74 前体可分别得到具有特定孔结构和高比表面积的Co@NC和Co@C核壳催化剂[78],Co@C催化剂中具有大孔径的碳壳既能保护Co 颗粒免于团聚失活又利于烃类扩散,因此具有较高的CO 转化率(30%)和C+5选择性(65%);Co@NC 催化剂的碳壳中富电子的N 可抑制烯烃的进一步加氢,使其对低碳烯烃具有较高的选择性(37%)。Fe@NC 核壳催化剂的富氮碳壳有利于CO2吸附,这使其在CO2加氢制低碳烯烃中的反应速率比基准Fe2O3纳米颗粒高出约25%,烯烃/烷烃比高出24 倍(图17)[79]。在没有K、Na 和S 等促进剂干扰的情况下,由MIL-101 热解生成Fe@C 催化剂中θ-Fe3C 是FTS 的活性相,具有比α-Fe 或由α-Fe 演化而来的铁碳化物更高的选择性和较低的CH4选择性。纳米粒子周围的多孔石墨烯壳层可抑制其聚集,从而提高催化剂的稳定性;而非晶态碳壳和石墨碳壳均不能有效保护催化剂免于氧化和颗粒聚集。在最优条件下,多孔石墨烯壳层包覆的Fe@C 催化剂对C+5选择性达57%~60%,对CH4选择性只有10%~12%[80]。
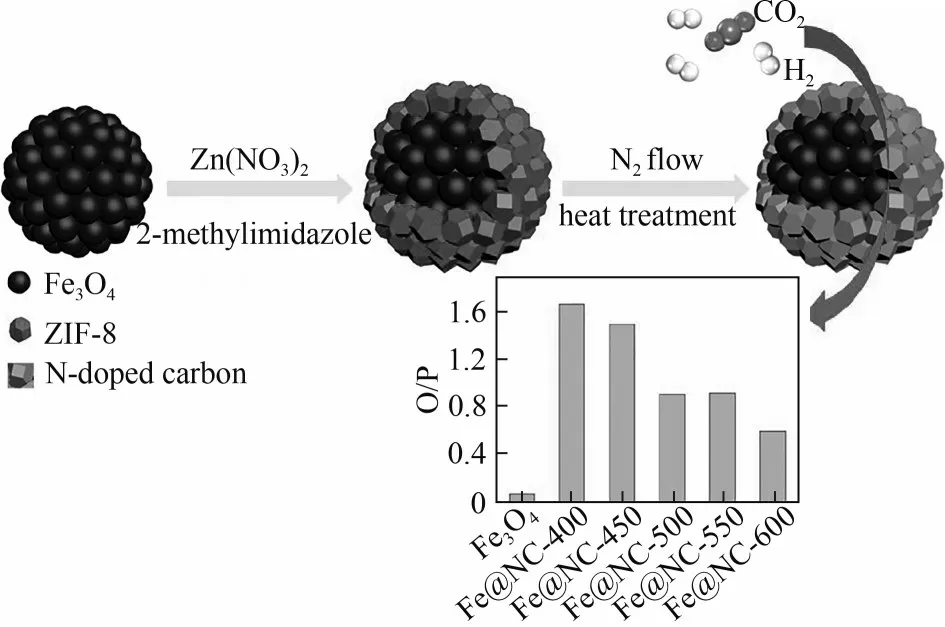
图17 Fe@NC催化剂的制备流程及不同催化剂上C2~C4的烯烃/烷烃值(O/P)[79]Fig.17 Preparation process of Fe@NC catalyst and olefin to paraffin ratio(O/P)of C2—C4 on different catalysts[79]
2.5 分子筛壳
分子筛的择形性和酸性使其具有较强的烃类聚合、异构化、芳构化和裂解能力[81],因此人们将分子筛和含氧中间体合成催化剂或费托催化剂耦合构建双功能催化剂,用于CO/CO2加氢制备高值化学品。影响这一接力反应进行的一个重要因素是含氧中间体合成催化剂或费托催化剂上的中间体向分子筛的扩散效率。常规负载型和物理混合型双功能催化剂提供的是一个开放和不受限制的反应环境,两类反应是随机和独立发生的。含氧中间体合成催化剂或费托催化剂上生成的中间体可以直接扩散到气相中,而不在分子筛上发生二次反应。相比较而言,以含氧中间体合成催化剂或费托催化剂为核、分子筛为壳的核壳催化剂可以提供一个受限的、一体化的催化环境,其中核与壳分别催化不同的反应。在核壳双功能催化剂中,核上形成的中间体必须通过分子筛壳扩散,在扩散到气相之前先在分子筛上进行二次反应,这种策略会增强核体初级反应产物向分子筛上酸性位点的迁移效率,从而提高目标产物的收率。
分子筛晶体中定向均匀排列的孔道和尺寸固定的孔径,决定了进出分子筛内部的分子大小。具有八元环结构、孔口尺寸为0.38 nm的SAPO-34分子筛被广泛用于CO/CO2加氢制低碳烯烃。在FTS 反应中,Fe3C@SiO2@SAPO-34 催化剂的SAPO-34 壳层微孔结构和独特酸度可使C+6的生成被完全抑制,C2~C4烃类成为主要产物(>50%)[82]。CuZnZr@SAPO-34催化剂的SAPO-34包覆可降低接触界面和金属位点的加氢能力,提高烯烃的选择性,并抑制CH4的生成。同时,Zn 的加入可降低壳层SAPO-34 的酸性,大幅度抑制初级烯烃的二次加氢反应,这些使CuZnZr@Zn-SAPO-34(4/1)催化剂对CO2加氢制低碳烯烃选择性高达72%[83]。
酸量较少、疏水性强和孔口尺寸小(0.446~0.47 nm)的Silicalite-1 分子筛(全硅型ZSM-5 分子筛)也常被用于CO/CO2加氢制低碳烃和低碳含氧化合物。在FTS 反应中,Fe/SiO2@Silicalite-1催化剂的壳层分子筛限域效应和择形性使产物中低碳烯烃选择性从20.22%提升到29.94%,而CH4和C+5的生成被抑制[84]。具有核壳结构的RhMn@S-1催化剂对合成气制C2-含氧化合物具有优异的催化效果,刚性沸石骨架稳定的Mn-O-Rhδ+结构是C2-含氧化合物生成的关键位点,最佳条件下CO 转化率为42.4%,产物中含氧化合物的总选择性达到40.3%,其中C2-含氧化合物的选择性达到88.3%,明显优于以前的催化剂(图18)。在长期试验中,RhMn@S-1催化剂表现出恒定的C2-含氧化合物产率和优越的耐久性[85]。

图18 RhMn@S-1的电子显微表征和不同RhMn基催化剂上C2-含氧化合物的平均产率及甲烷和CO2选择性[85]Fig.18 Electronic microscopic characterization and average productivities of C2-oxygenates and selectivities of methane and CO2 over various RhMn-based catalysts[85]
ZSM-5 分子筛骨架具有二维双十元环孔道结构,孔口尺寸为0.51~0.56 nm,可调节的酸量使其对烯烃齐聚、异构化和芳构化反应有着重要作用。在FTS 制异构烃反应中,Co/SiO2@H-ZSM-5 催化剂的壳层分子筛包覆保证了Co/SiO2催化剂上生成的长链烃全部经过分子筛壳层,并在其酸性位点上发生裂解和异构反应,产物中烃类的生成完全被抑制[86-87]。随着Co/SiO2@H-ZSM-5 催化剂的核体Co/SiO2粒径减小,异构烃与正构烃的比、烯烷比均逐渐提高,但CH4的选择性也有小幅上升[88]。在相同的水热合成条件下,仅通过减小沸石胶囊催化剂核的尺寸即可实现壳层厚度的增加。随着Ru/SiO2@HZSM-5 催化剂中核体粒径的减小和分子筛壳层厚度的增加,FTS 产物中异构烷烃的选择性会逐渐提高,且明显高于Ru/SiO2及其与H-ZSM-5 物理混合催化剂的结果[89]。在Co/SiO2@H-ZSM-5 的制备过程中,改变模板剂和水热条件可对壳层分子筛的厚度和酸性特征进行调变,进而改变产物中异构烷烃的选择性[90]。相比于传统铁基催化剂和机械混合催化剂,H-ZSM-5 包覆熔铁的核壳催化剂既可降低FTS 中CH4和CO2的选择性又可明显提高异构与正构烷烃的比例[91]。Cu/ZnOx(1.38)@Na-ZSM-5催化剂的壳层分子筛限域效应可有效抑制Cu-ZnOx相界面分离,最优条件下Cu/ZnOx(1.38)@Na-ZSM-5 核壳催化剂对甲醇的时空收率[44.88 g MeOH/(g Cu·h)]远高于相同条件下负载型Cu/ZnOx/Na-ZSM-5 催化剂[13.32 g MeOH/(g Cu·h)]和工业Cu/ZnO/Al2O3催化剂[8.46 g MeOH/(g Cu·h)]上的结果,且该催化剂在长期试验中具有恒定且优越的甲醇时空收率(图19)[92]。FeMn@Hollow HZSM-5 核壳催化剂的时空有序效应可充分发挥HZSM-5分子筛的择形效应和芳构化能力,使FTS 产物中芳烃的时空产率高达1.9 g/(g Fe·h),突破了以往催化剂的芳烃时空产率不超过1.0 g/(g Fe·h)的瓶颈,该催化剂的核体元素组成、连接介孔孔道的中空结构以及可调的纳米反应器酸碱度对提高芳烃的产量和抑制积炭具有重要作用(图20)[8]。

图19 不同催化剂的CO2加氢反应性能和Cu/ZnOx(1.38)@Na-ZSM-5催化剂在CO2加氢反应中的稳定性[92]Fig.19 CO2 hydrogenation performance of different catalysts and durability of the Cu/ZnOx(1.38)@Na-ZSM-5 catalyst in CO2 hydrogenation[92]

图20 FeMn@Hollow HZSM‐5催化剂的催化性能[8]Fig.20 Catalytic performance of FeMn@Hollow HZSM‐5 catalyst[8]
具有三维十二元环交叉孔道结构、孔口尺寸为0.56~0.77 nm 的高硅Beta 型分子筛对烃类异构反应具有较好的催化活性。相比于Co/Al2O3和Co/Al2O3与Beta 机械混合的催化剂,β-zeolite/Co/Al2O3催化剂(图21)的特殊核壳机构可增加中间体和活性位点之间的碰撞概率,从而提高中间异构烃的选择性及异构烷烃与正构烷烃的比例,同时,烃类的生成被完全抑制,副产物CH4选择性也较低(表2)[93]。采用TEAOH 对Co/Al2O3回流预处理有利于FTS 催化剂表面清洁和颗粒内部孔径的增大,在包覆前用乙醇浸泡Co/Al2O3可有效避免内部孔道的破坏以及Co物种向Beta 分子筛的迁移,得到纯净的、均匀的Hβ 分子筛包覆Co/Al2O3的核壳双功能催化剂。在FTS 反应中,该核壳催化剂上异构烷烃/正构烷烃的摩尔比较物理混合的催化剂提高了约64%[94]。

表2 常规Co/Al2O3、Co/Al2O3和Beta沸石物理混合物及沸石包覆Co/Al2O3催化剂的催化性能对比[93]Table 2 Comparison of the catalytic performance of conventional Co/Al2O3,a physical mixture of Co/Al2O3 and Beta-zeolite,and the zeolite-coated Co/Al2O3 catalyst[93]
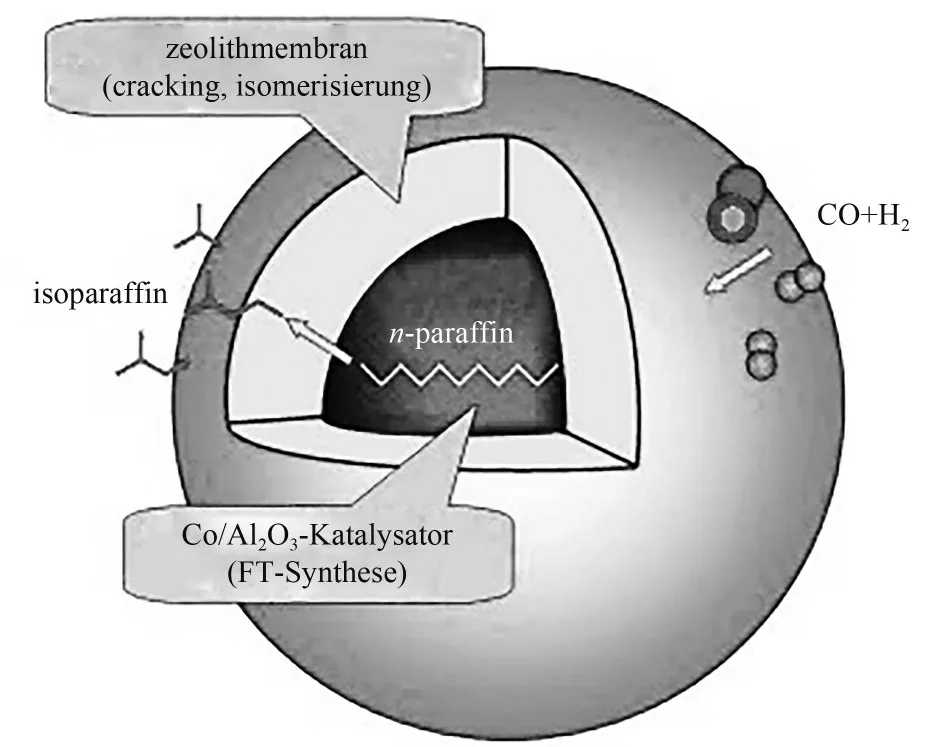
图21 β-zeolite/Co/Al2O3核壳催化剂[93]Fig.21 β-zeolite/Co/Al2O3 core-shell catalyst[93]
兼具十二元环直通孔道0.65 nm×0.70 nm 和八元环直通孔道0.26 nm×0.57 nm 的MOR 分子筛被广泛应用于长链烃类的异构化和裂解重整反应。在FTS 制乙醇反应中,Cu/ZnO@H-MOR 胶囊催化剂的空间受限自调控机制可实现接力催化,抑制副反应发生。同时,通过改变H-MOR 分子筛壳内Brønsted酸性位点的数量,可调节分子筛胶囊催化剂的活性和乙醇的选择性[95]。
双壳层包覆的Cr/ZnO@Silicalite-1@H-ZSM-5催化剂上进行的接力催化反应还可实现合成气到二甲醚的精确控制合成,同时抑制了其他副产物的生成[98]。Wang 等[99]为CO2加氢制备异构烃反应设计了一系列以Fe-Zn-Zr 为核,以HZSM-5、HBeta、HY分子筛为壳层的核壳催化剂,它们对异构烷烃的选择性远高于Fe-Zn-Zr 与沸石机械混合的催化剂。HZSM-5 与HBeta 分子筛的协同作用还可使Fe-Zn-Zr@HZSM-5-HBeta(4∶1)催化剂表现出超高异构与正构烷烃比(81.3%)。此外,包覆前用TPABr 水溶液对Fe-Zn-Zr 氧化物进行高温水热处理,可抑制FTS 反应发生,使CO2尽可能多地转化为HCOO*和CH3O*中间体,并在HZSM-5 的强Brønsted 酸位上进行齐聚和异构化反应(图22)。优化后Fe-Zn-Zr(0.1∶1∶1)-T-24h@HZSM-5 催化剂上生成的汽油中C+5异构烷烃的选择性达到93%,而副产物CO 的选择性只有24%[100]。

图22 CO2加氢制优质汽油复合催化剂上不同氧化物表面物种可能的演化及反应机理[100]Fig.22 Possible evolution of surface species on different oxides and reaction mechanism on composite catalysts for CO2 hydrogenation to high-quality gasoline[100]
不同分子筛的酸性和孔径大小各不相同,将不同分子筛与RWGS/FTS催化剂耦合构建核壳双功能催化剂可实现CO/CO2加氢制备特定高值化学品。然而,核壳双功能催化剂中邻近效应也是影响CO/CO2加氢催化活性的一个重要因素,解析邻近效应来提高接力催化的效率是后续研究的一个重难点。
3 结语与展望
核壳纳米材料作为CO/CO2热催化加氢反应的催化剂具有众多优势。金属单质壳和分子筛壳包覆的核壳结构可灵活地在同一催化剂中引入不同的催化活性位点,实现接力催化。金属氧化物壳包覆可改变核体催化剂的电子效应和表面碱度。金属氧化物、SiO2、C 和分子筛壳包覆可抑制核体金属原子、团簇或纳米颗粒的团聚烧结,从而大幅度提高催化剂的活性和寿命。核壳结构还可提供一个受限的、一体化的催化环境,提升核体初级反应产物向壳层活性位点的迁移效率,从而提高催化活性和目标产物的选择性。需要指出的是,多种壳层包覆的结合可使核壳催化剂的稳定性和催化性能更佳。
由于核壳结构的构造方式以及组合策略各异,所以核壳纳米材料的制作方法十分丰富。本文介绍了核壳纳米材料的常用制备方法,包括溶胶-凝胶法、水/溶剂热法、微乳液法、化学气相沉积法、层层组装法、热分解法和机械球磨法等。重点介绍了它们的基本原理、可控合成的关键因素及优缺点。需要指出的是,多种方法的结合可有效控制制备过程,获得性能更好、更有针对性的核壳材料。
目前,核壳催化剂在CO/CO2热催化加氢反应中的应用还处于初期,为了实现CO/CO2高活性、高选择性地转化为特定产物,在核壳催化剂合成策略、传质效应、反应机理和构效关系等方面还需要深入研究。当前在CO/CO2热催化加氢反应中的应用已证明调控核壳催化剂的设计变量可改变反应体系内的传热、传质、本征反应活性和接力催化效果。除此之外,结合实际条件开发简单、快捷和具有经济竞争力的核壳纳米催化剂大规模合成新工艺也是未来商业化的必经之路。
