超分子前体制备g-C3N4/g-C3N4同质结及光催化性能研究
2022-03-11班昌胜左龙涛
班昌胜,李 军,金 央,陈 明,左龙涛
(四川大学化学工程学院,四川成都 610065)
随着全球工业化的发展,印染废水产生的环境污染问题日趋严重[1]。此类有机污染物毒性大、色度高,且抗生物降解性强,因此迫切需要开发高效的染料废水处理技术。光催化降解技术由于其可循环性、无二次污染等特点,被认为是最具有发展前景的染料废水处理技术之一[2]。
在众多的光催化材料中,氮化碳(g-C3N4)具有原料廉价易得、物理化学稳定性较高、可见光响应良好等优势,因此近年来受到了广泛的研究[3]。然而以三聚氰胺等常规含氮前驱体制备的g-C3N4通常是块体材料,这主要是因为三聚氰胺的熔点在250℃,而三聚氰胺生成三嗪环并进一步聚合的温度在350℃以上,所以前驱体转变为g-C3N4之前处在熔融状态,从而导致制备的g-C3N4结构致密性很强,即比表面积较低(约10 m2/g)[4]。三聚氰胺-三聚氰酸(MCA)超分子,由于其内部氢键的存在在g-C3N4的生成温度下仍能保持结构的稳定性,以其为前驱体一步煅烧即可制备比表面积较高的g-C3N4,同时保留部分前驱体的特征形貌[5]。然而纯相的g-C3N4在光催化应用中依然受到光生空穴-电子对复合率较高的限制。许多研究工作通过构造异质结有效提升了电荷的分离效率,但是构造高效异质结必须同时考虑各种因素,包括晶体结构、结晶度、表面性质等的差异。相比之下,同质结由于其相似的电子结构单元,更有利于载流子的分离与迁移,因此同质结界面的有效接触是提升电荷分离效率的关键[6]。一般来说,将两种或多种g-C3N4原料混合后煅烧可有效提升两相界面的接触程度,但三聚氰胺、二氰二胺、尿素等原料会在高温下熔融继而相互溶解,最终生成均一结构的g-C3N4,也就无法因两相费米能级的差异形成内电场从而加速光生载流子的分离[7]。而MCA超分子的高热稳定性使其在煅烧过程中仍能保持相对独立的结构,并且不同方法制备的超分子前驱体煅烧得到的g-C3N4在能带结构上表现出差异[8],以此为基础制备的同质结光催化剂可以通过拓展π-π共轭体系实现载流子的高效传输。
因此,笔者分别以二氰二胺水热法和三聚氰胺-三聚氰酸共沉淀法制备了MCA超分子,一步煅烧混合前驱体制备了兼具多孔纳米片和中空纳米管形貌的g-C3N4/g-C3N4同质结。通过模拟可见光照射下降解水相中的亚甲基蓝测试材料的光催化效率,考察了不同前驱体比例制备的同质结的表面性质及催化效率的差异,通过自由基捕获实验和X射线光电子能谱价带谱测试对同质结的光催化机理进行分析。
1 实验部分
1.1 原料和仪器
原料:二氰二胺、三聚氰胺、三聚氰酸、亚甲基蓝(MB)、异丙醇、对苯醌、乙二胺四乙酸二钠、盐酸、乙醇,均为分析纯。
仪器:OTF-1200X高温管式反应炉;PCX-50B光化学反应仪;H1850高速离心机;UV-1100紫外-可见分光光度计。
1.2 材料制备
1.2.1 水热法MCA的制备
将30 g二氰二胺溶解于60℃的150 mL二级水中,趁热将其转入200 mL水热反应釜中,在160℃水热反应8 h。反应完成后自然冷却至室温,得到的白色粉末用二级水离心洗涤3次,在80℃干燥10 h得到水热法MCA超分子,记为MCA-H。
1.2.2 沉淀法MCA的制备
将2.522 g三聚氰胺和2.582 g三聚氰酸分别溶解于80℃的100 mL二级水中,将三聚氰胺溶液缓慢滴加到三聚氰酸溶液中,得到白色乳液并持续搅拌2 h,在80℃蒸干水分得到共沉淀法MCA超分子,记为MCA-P。
1.2.3g-C3N4/g-C3N4同质结的制备
将质量比分别为1∶3、1∶1、3∶1的MCA-H和MCA-P混合使总质量为2 g,研磨后放入磁舟中,置于管式炉中在Ar气保护下于550℃煅烧2 h,升温速率为10℃/min,得到淡黄色蓬松粉末即g-C3N4/g-C3N4同质结,分别记为CN-HP-1/3、CN-HP-1/1、CN-HP-3/1。将2 g的MCA-H和MCA-P分别以同样的条件煅烧得到单体g-C3N4,分别记为CN-H和CN-P。为对比将2 g三聚氰胺在550℃煅烧4 h得到块体g-C3N4,记为BCN。
1.3 材料表征
采用XRD-6100型X射线衍射仪(XRD)分析样品的晶型。采用JSM-7500F型扫描电镜(SEM)和Tecnai G2 F20 S-TWIN型透射电镜(TEM)观察样品的形貌。采用Nicolet iS50型傅里叶变换红外光谱仪(FT-IR)对样品的特征官能团晶型进行分析。采用AXIS-Supra型X射线光电子能谱(XPS)对样品的化学态进行分析,以碳标准结合能(284.6 eV)对高分辨率数据进行矫正,同时进行了XPS价带谱测试。采用ASAP2460比表面积分析仪对样品进行氮气吸附-脱附测试,并使用Barret-Joyner-Halenda(BJH)计算材料的比表面积及孔径分布,p/p0=0.98时的吸附体积用于确定孔体积。半导体材料经紫外光照射,部分激发态的电子和空穴复合后会将部分能量以荧光的形式发射出来,测试光致发光光谱(PL)以表征样品的载流子复合程度,测试仪器为F-700型荧光分光光度计,激发光波长统一采用287 nm。
1.4 光催化性能测试
通过模拟可见光降解MB溶液的效率来测试材料的光催化性能,测试仪器为PCX-50B Discover多通道光催化反应系统。将20 mg光催化剂超声分散于40 mL质量浓度为10 mg/L的MB溶液中,暗光搅拌30 min达到吸附平衡。开始光照后每15 min取3 mL溶液离心分离,离心时间为5 min,转速为8 000 r/min,取上清液测量吸光度并记录。
为探究催化过程产生的活性物种及反应机理,分别以对苯醌(BQ)、异丙醇(IPA)、乙二胺四乙酸二钠(EDTA-2Na)为超氧自由基(·O2-)、羟基自由基(·OH)、光生空穴(h+)的捕获剂进行捕获实验,投加量为0.01 mol/L。
2 结果与讨论
2.1 晶型分析
图1为不同方法制备的MCA(a)和单体g-C3N4及CN-HP同质结(b)的XRD谱图。从图1a看出,MCA超分子在2θ为10.6、18.3、27.8°处的晶体衍射峰分别对应MCA的(100)(110)(002)晶面。水热法MCA是由二氰二胺在高温高压下部分聚合生成三聚氰胺并放出氨气导致反应体系pH增大,生成的三聚氰胺部分水解生成三聚氰酸,三聚氰胺和三聚氰酸自组装得到MCA超分子;共沉淀法MCA是将等物质的量的三聚氰胺和三聚氰酸溶解于热水中,通过常压混合、结晶沉淀得到MCA超分子。因此,共沉淀法MCA含有没有完全反应原料的特征峰,而水热法MCA是在高温高压下合成因此结晶度更高,这对应于MCA-H衍射峰强度较MCA-P更高。另外,经过测量水热法MCA的密度是沉淀法MCA密度的2倍以上,从侧面证明两种前驱体结晶结构具有差异。
从图1b看出,单体g-C3N4及CN-HP同质结在2θ为12.8、27.3°处出现的2个衍射峰分别对应g-C3N4的(100)(002)晶面(JCPDS 87-1526)[9]。与CN-H相比,CN-P的(002)衍射峰向更高角度偏移,说明其共轭片层间的堆叠更为紧密。CN-P在2θ为21.1°处还存在一个很弱的宽峰,CN-H则不存在这个峰。当三聚氰胺在较低的温度下(500℃以下)煅烧时也会出现这个峰。这是因为低温抑制了melon结构的聚合。这说明CN-H的聚合度更高,这主要源自于水热法MCA的致密结构[10]。此外,CN-HP-3/1和CN-HP-1/1在2θ为31.4°处出现一个不常见的小尖峰,这可能是MCA-H和MCA-P不完全共聚合导致的。

图1 不同方法制备的MCA(a)、单体g-C3N4及CN-HP同质结(b)的XRD谱图Fig.1 XRDpatterns of MCA prepared by different methods(a),monomer of g-C3N4 and CN-HPhomojunctions(b)
2.2 形貌分析
图2为CN-H(a~b)、CN-P(c~d)、CN-HP-1/1(e~f)的SEM照片和CN-HP-1/1(g~h)的TEM照片。从图2a~b看出,CN-H具有堆叠的3D纳米片结构,这主要是因为在流动Ar气的吹扫作用下,均三嗪环层间连接的氨基更容易脱除为氨气,从而导致层间分离度增大,出现多孔片状堆叠的形貌。从图2c~d看出,CN-P具有不规则的中空管状结构,这主要是由于沉淀法MCA还有部分未完全反应的三聚氰胺或三聚氰酸,在煅烧过程中棒状的MCA外层温度迅速升高并快速热聚合形成均三嗪环结构,热稳定性增强;而内层温度受传热限制提升较慢,因此在形成均三嗪环之前会形成熔融态并大量气化,从而形成不规则的中空管状形貌。从图2e~f看出,同质结CN-HP-1/1兼具多孔片状及中空管状结构,这证明了MCA-H和MCA-P并未在高温下熔融聚合为单一结构的g-C3N4,同时可以看出中空管状结构的中空程度进一步增强,这是因为水热法MCA羟基含量较高,热聚合过程中会放出大量水蒸气,而水蒸气会带走更多的气化组分,促进中空管的形成和破裂,这也为催化反应提供了更多的活性位点。图2g~h进一步显示出CN-HP-1/1具有中空管状和纳米片的双重结构,然而在高分辨率透射电镜照片中并未出现清晰有序的晶格条纹,这可能是因为CN-HP同质结是多晶和非晶的混合态[11]。

图2 CN-H(a~b)、CN-P(c~d)、CN-HP-1/1(e~f)的SEM照片和CN-HP-1/1(g~h)的TEM照片Fig.2 SEMimagesof CN-H(a~b),CN-P(c~d),CN-HP-1/1(e~f);TEMimagesof CN-HP-1/1(g~h)
为进一步探究材料的形貌差异,对其进行了BET法比表面积测试。表1为样品的比表面积、平均孔径和孔容。从表1看出,MCA超分子制备的g-C3N4较块体g-C3N4比表面积有显著提升,同质结的比表面积较单体略有减小,这可能是因为部分破碎的中空管插入到纳米片层间的空隙中,使得比表面积和孔容有所减小。
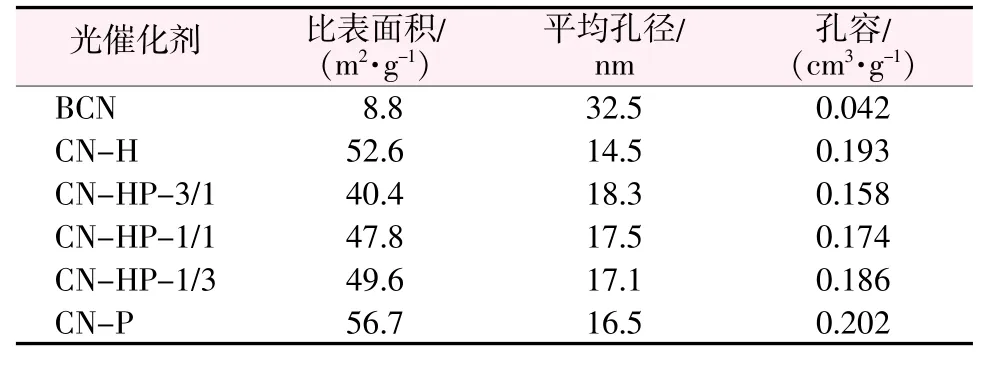
表1 光催化剂样品的比表面积、平均孔径及孔体积Table 1 Specific surface area,average pore diameter and pore volumeof samples
2.3 化学键分析
图3为同质结(CH-HP-1/1)及单体氮化碳(CN-H、CN-P)的XPS图。从图3a看出,在3种样品的XPS全谱图上均检测出C、N、O3种元素。从图3b看出,在C 1s高分辨率XPS图上,CN-H、CN-HP-1/1、CN-P的杂质C结合能峰均出现在284.6 eV处,CN-H和CN-P的N—C=N键结合能为287.9eV,而CN-HP-1/1的N—C=N键结合能减小至287.8eV,这可能是界面间电荷转移的结果。值得注意的是,CNHP-1/1的C—C与N—C=N峰强度之比为0.24,分别高于CN-H(0.10)和低于CN-P(0.26),说明水热法前驱体制备的CN-H杂质C含量更低,同质结的杂质含量位于两单体之间。从图3c看出,样品的N 1s高分辨率XPS图中位于398.3、399.7、400.9 eV的3个峰分别对应于C=N—C、N—[C]3、C—N—H[12]。同样值得注意的是,CN-HP-1/1的C—N—H与C=N—C峰强度之比为0.11,而CN-H和CN-P的峰强度之比分别为0.12和0.13,说明CN-H和CN-P之间表面耦合产生了强相互作用,这在很大程度上要归公于π-π共轭化学键结构的相似性,以此构建的同质结有效促进了电荷的分离与转移[13]。
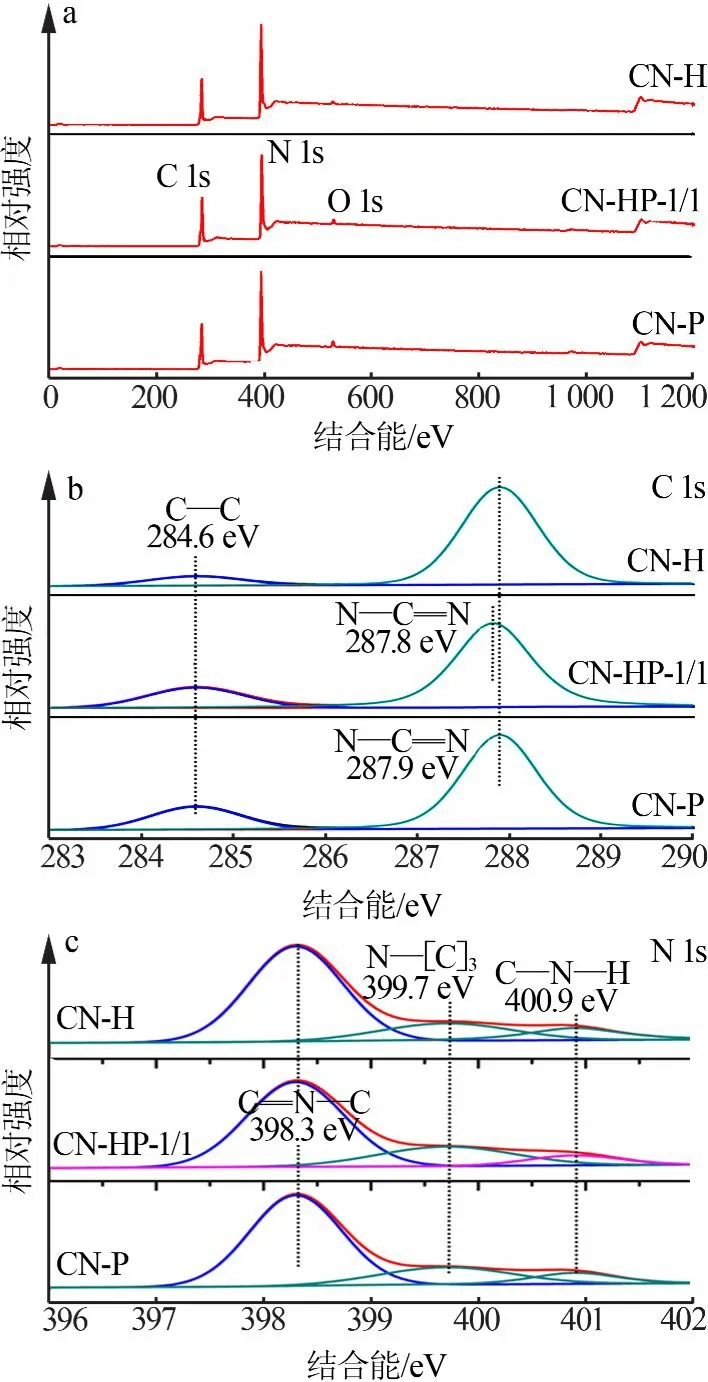
图3 同质结(CN-HP-1/1)及单体氮化碳(CN-H、CN-P)的XPS图Fig.3 XPSspectra of homogeneous junction(CN-HP-1/1)and monomer of CN(CN-H,CN-P)
2.4 光催化测试
图4为光催化剂样品可见光催化降解MB的性能测试结果(a为MB相对浓度随时间的变化;b为伪一级动力学常数;c为循环性能图;d为光催化反应前后CN-HP-1/1的XRD谱图)。从图4a、b看出,CN-HP-1/1的催化效果最好,其伪一级反应常数为块体g-C3N4的5倍,是CN-H和CN-P的1.6倍左右。值得一提的是,CN-HP-3/1的催化性能较CN-H和CN-P有所下降,可能是由于样品在热聚合过程中特定比例的前驱体结构出现坍塌,导致了结构缺陷。从图4c、d看出,5次循环实验后CN-HP-1/1对MB的降解率仍在99.0%以上,并且反应前后样品的XRD谱图几乎没有变化,证明了同质结结构的稳定性。
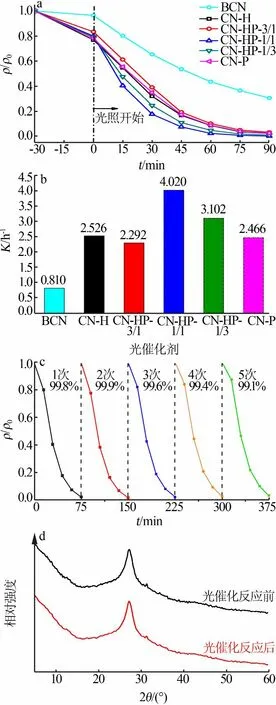
图4 光催化剂可见光催化降解MB的性能Fig.4 Visible light catalytic degradation performance test of methylene blue
为排除比表面积对同质结光催化性能提升的影响,以伪一级动力学常数除以各光催化剂的比表面积得到单位面积反应速率常数,结果见图5。从图5看出,CN-HP-1/1同质结单位面积的反应常数较单体提升1.8倍左右,说明同质结的构建有效提升了光催化剂单位面积的光催化活性。
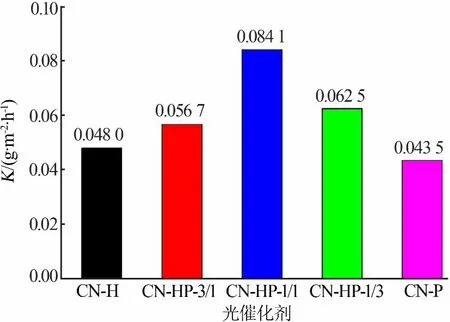
图5 光催化剂单位比表面积的反应速率常数Fig.5 Reaction rate constant per unit specific surface area
2.5 光化学测试
为探究同质结的光化学性能,对材料进行了紫外-可见漫反射吸收光谱(a、b)及光致发光光谱(c)测试,结果见图6。从图6a看出,CN-H、CN-P以及同质结的光吸收边缘位置区别不大,很大程度上是源于水热法MCA和共沉淀法MCA本征结构的相似性。同质结光催化剂较CN-H和CN-P对紫外光的吸收强度呈现出差别,其中CN-HP-1/1和CN-HP-1/3的吸收强度最高,而CN-H的吸收强度最低,CN-P次之,这是因为单体g-C3N4间的有效连接拓展了π-π共轭体系在同质结中的占比,因而增强了光谱吸收能力。通过延长线的截距求得材料的禁带宽度(见图6b),CN-H、CN-HP-3/1、CN-HP-1/1、CNHP-1/3、CN-P的禁带宽度分别为2.72、2.78、2.76、2.78、2.80 eV。同质结催化剂的禁带宽度均处于单体CN-H和CN-P之间,说明了两种单体在复合材料中的有机结合。
对光催化剂进行了光致发光光谱测试以表征材料的空穴-电子对复合率(见图6c)。从图6c看出,与CN-H和CN-P相比,CN-HP-1/1和CN-HP-1/3的荧光强度更低,对应于更低的光生载流子复合率,说明同质结间有效电荷传输通路的构建。然而CNHP-3/1的荧光强度比单体g-C3N4更高,这与其催化降解MB的效率较低相印证,并且其荧光强度峰同块体g-C3N4相似,证明了该比例下前驱体高温熔融态的出现。CN-HP-1/1的荧光发射峰较CN-H和CN-P出现了红移,这是两相紧密的堆叠结构导致比表面积下降的结果。
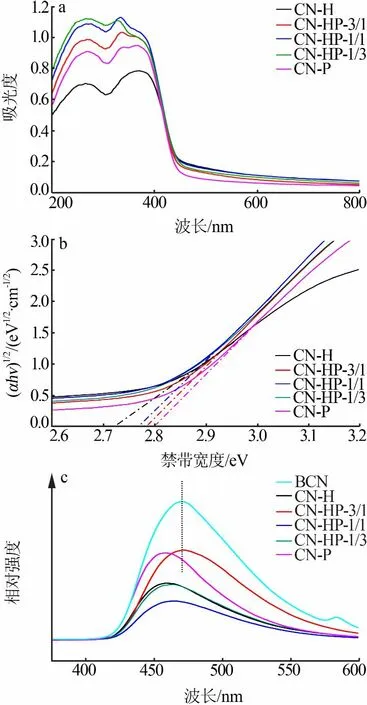
图6 光催化剂紫外-可见漫反射吸收光谱(a)、禁带宽度(b)和光致发光光谱(c)Fig.6 UV-visabsorption spectra(a),band gap(b),photoluminescence spectra of photocatalyst(c)
2.6 光催化机理分析
为探究同质结的构建对光催化活性自由基团氧化还原能力的影响,对单体g-C3N4和同质结光催化剂进行了自由基捕获测试,分别以对苯醌(BQ)、异丙醇(IPA)、乙二胺四乙酸二钠(EDTA-2Na)捕获超氧自由基(·O2-)、羟基自由基(·OH)、光生空穴(h+),结果见图7a。从图7a看出,异丙醇的加入对MB的降解抑制作用很小,这与g-C3N4光生空穴的氧化能力不足以分解水产生强活性的羟基自由基相符。对苯醌和EDTA-2Na的加入都较大程度地抑制了MB的降解,但不同的是EDTA-2Na的加入对CN-HP-1/1的抑制作用较单体g-C3N4更强,说明同质结较单体g-C3N4的价带位置出现了变化。
为进一步探究能带位置的变化,XPS价带谱测试结果见图7b。从图7b看出,CN-H、CN-HP-1/1、CN-P价带顶位置相对于费米能级(EF)分别处于约2.0、2.2、2.1 eV。氢还原电势(H+/H2)相对于真空度约为4.5 eV,g-C3N4的功函数为4.0 eV[14],因此CN-P和CN-H的价带位置相对于标准氢电极(vs.NHE=0)分别处于1.5、1.6eV,计算出导带位置分别为-1.22eV和-1.20 eV。相对于单体g-C3N4,同质结CN-HP-1/1的价带处于更正的位置,即受光激发产生的光生空穴的氧化性能更强。Ⅱ型电子传输结构最大的特点是牺牲氧化还原能力以增强空穴-电子对的分离效率,而CN-HP同质结在抑制载流子复合的同时保留了空穴的氧化能力,说明同质结催化剂更符合Z型电子传输机制。电荷传输机理见图8。从图8看出,CN-H导带上的光生电子同CN-P价带上的空穴复合,留下CN-H价带上氧化能力更强的光生电子直接氧化降解MB,而CN-P导带上的还原能力更强的电子同溶解氧反应生成活性更高的超氧自由基后参与MB的降解。

图7 自由基捕获实验结果(a)和XPS价带谱测试结果(b)Fig.7 Free radical capture experiment(a),XPS valence band gap spectra test(b)
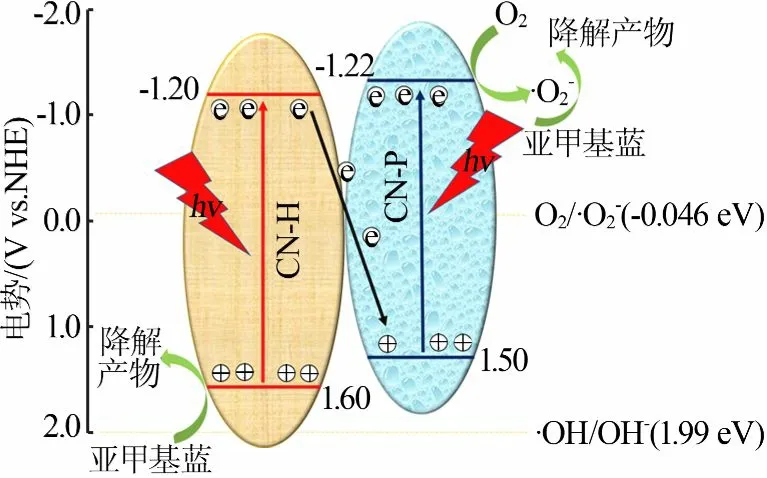
图8 CN-HP同质结光催化示意图Fig.8 Photocatalytic mechanismof CN-HPhomojunctions
3 结论
使用二氰二胺水热法和三聚氰胺-三聚氰酸共沉淀法分别合成了MCA超分子,以混合超分子为前驱体一步煅烧制备了兼具多孔纳米片和中空纳米管形貌的g-C3N4/g-C3N4同质结。MCA超分子内部氢键结构的高热稳定性使其不会在热聚合过程中熔融,保留了两相g-C3N4在同质结内的差异性。同质结光催化剂在MB降解中表现出良好的光催化性能和循环稳定性,其中CN-HP-1/1降解速率最高,为块体g-C3N4的5倍。光致发光光谱测试表明,具有合适比例的MCA制备的同质结光催化剂的光生电子-空穴复合率较单体进一步降低。自由基捕获实验和XPS价带谱测试表明,同质结价带位置较单体g-C3N4更正,光催化降解MB过程中起主导作用的是光生空穴,分析表明同质结具有Z型电子传输通路。该工作为一步煅烧法制备多孔石墨相氮化碳同质结提供了可行的思路。
