二维硼氮Kagome材料的力学行为和电子性质研究
2022-03-09向燕宁冯振李发闯郭朝博杨泽林方苗苗
向燕宁 冯振 李发闯 郭朝博 杨泽林 方苗苗

摘 要:Kagome晶格因包含平带和狄拉克锥引起了人们的广泛关注。采用基于密度泛函理论的计算方法,以二维硼氮Kagome材料为研究对象,系统研究了硼氮Kagome材料的原子结构、力学性质、电子结构以及对氮气分子的吸附作用。结果表明:硼氮Kagome材料的化学式为B6N6,晶格常数为9.97 Å,测量B—B、B—N、N—N的键长分别为1.64 Å、1.33 Å、1.19 Å;电荷分析表明B—N为离子键,而N—N和B—B为共价键;该二维材料展示出各向异性的力学性能和无磁性的金属特性,其电子能带中存在两套Kagome能带;该材料具有较大孔洞结构,表现出对氮气分子的优异气敏特性。研究结果为硼氮Kagome材料在气体传感器件中的应用提供了理论参考。
关键词:二维材料;硼氮Kagome;力学性质;电子结构
中图分类号:O469 文献标志码:A 文章编号:1003-5168(2022)2-0093-05
DOI:10.19968/j.cnki.hnkj.1003-5168.2022.02.022
Mechanical Property and Electronic Structure of Two-Dimensional BN-Kagome
XIANG Yanning 1 FENG Zhen 1,2 LI Fachuang 1 Guo Chaobo 1 YANG Zelin 1
FANG Miaomiao 1
(1. School of Materials Science and Engineering, Henan Institute of Technology, Xinxiang 453003,China;
2.School of Physics, Henan Normal University, Xinxiang 453007,China)
Abstract: Kagome lattice has attracted much attention because it contains flat and Dirac bands. The atomic structure, mechanical properties, electronic structure and nitrogen adsorption of two-dimensional (2D) BN-Kagome materials were systematically studied by density functional theory. The results show that the chemical formula of BN-Kagome material is B6N6, and the lattice constant of B6N6 is 9.97 Å. The bond lengths of B—B、B—N and N—N are 1.64 Å,1.33 Å and 1.19 Å, respectively. At the same time,charge analysis shows that B—B is ionic bond,while N—N and B—B are covalent bond. The 2D BN-Kagome exhibits anisotropic mechanical property, non-magnetic metallic property,and two sets of Kagome bands. The 2D BN-Kagome has a large pore structure and has a low adsorption capacity for nitrogen molecules. Our results provide a theoretical reference for the application of BN-Kagome materials for gas sensors.
Keywords: two-dimensional materials; BN-Kagome; mechanical property; electronic structure
0 引言
近年來,具有单个或几个原子层厚度的二维材料引起了人们的广泛关注,在物理、材料、电子、能源等领域展示出巨大的发展潜力。二维材料的原子结构可以呈现出不同的晶格形式,如六角晶格、三角晶格和Kagome晶格。Kagome晶格是由共角对顶的三角格子组成,将蜂窝六角结构最近相邻的每个边的中心点连接起来可获得Kagome晶格,因此Kagome晶格可由蜂窝六角晶格变化而来。
Kagome晶格可容纳不同类型的量子态,根据紧束缚模型可知,它满足在相对论中粒子能量与动量关系的狄拉克方程,由此可知它包含狄拉克锥和无色散的平带,两者即为Kagome能带[1]。狄拉克锥可产生载流子迁移率极高的无质量费米子,这些无质量费米子能够在材料当中自由地运动形成弹道输运,而平带中包含质量无穷大的重费米子,平带意味着态密度非常大,这两者相结合使得系统中电子与电子之间的关联性非常强,即实现重费米子和轻费米子共存。Kagome能带会引起铁磁性、超导、Wigner晶格、拓扑电子态和电声耦合等。
麻省理工学院Checkelsky等人研究了d电子Kagome金属Fe3Sn2,观察到一个持续高于室温的固有异常霍尔电导率和处于费米能级附近的一对带隙为30 meV准二维狄拉克锥[2]。上海微系统所刘中灏等人利用角分辨光电子能谱、扫描隧道显微镜和理论计算对Kagome层状3d过渡金属CoSn进行了研究,直接观测到紧束缚模型预测的“教科书式”的Kagome特征能带结构,并且发现其能带结构具有轨道选择特性,为Kagome晶格电子能带及其轨道物理提供了一个典型范例[3]。
笔者在已有文献的基础上,采用第一性原理方法对BN-Kagome材料进行系统研究。分析原子结构特点,考察化学成键特性,测试力学行为,并研究电子结构特点,从而为其应用提供基本参考。
1 方法和参数设置
采用基于自旋极化的密度泛函理论Vienna Ab initio Simulation Package(VASP)软件包来模拟材料的相关性质[4]。采用投影缀加平面波方法处理离子实和价电子之间的交换关联作用,交换关联泛函为Generalized Gradient Approximation-Perdew Burke Ernzerof (GGA-PBE)[5]形式的广义梯度近似。经过测试,平面波截断能选用为500 eV,为避免二维层状材料各层之间的相互作用,真空层设定为20 Å。采用基于Grimme的DFT-D2方法描述二维材料与小分子之间的弱范德瓦耳斯力。计算时力和能量的收敛判据分别为0.01 eV·Å-1和1.0×10-5 eV。K点采用以第一布里渊区Gamma为中心的7×7×1网格进行结构弛豫和电子结构计算。计算结果采用Python编写的程序以及VASPKIT程序处理[6]。
2 结果与讨论
2.1 原子结构和稳定性
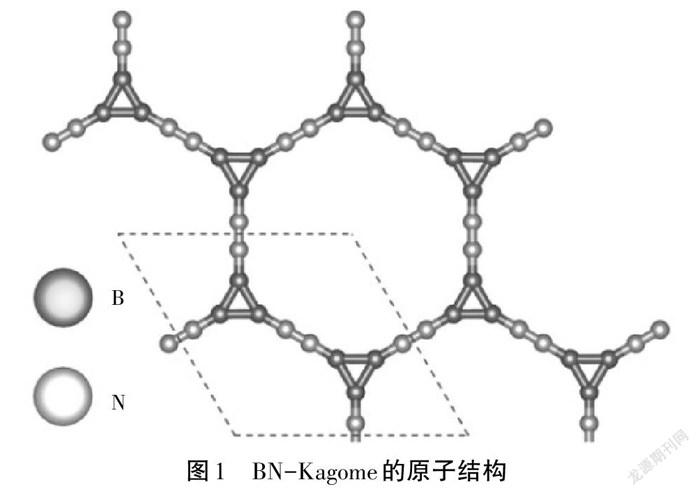
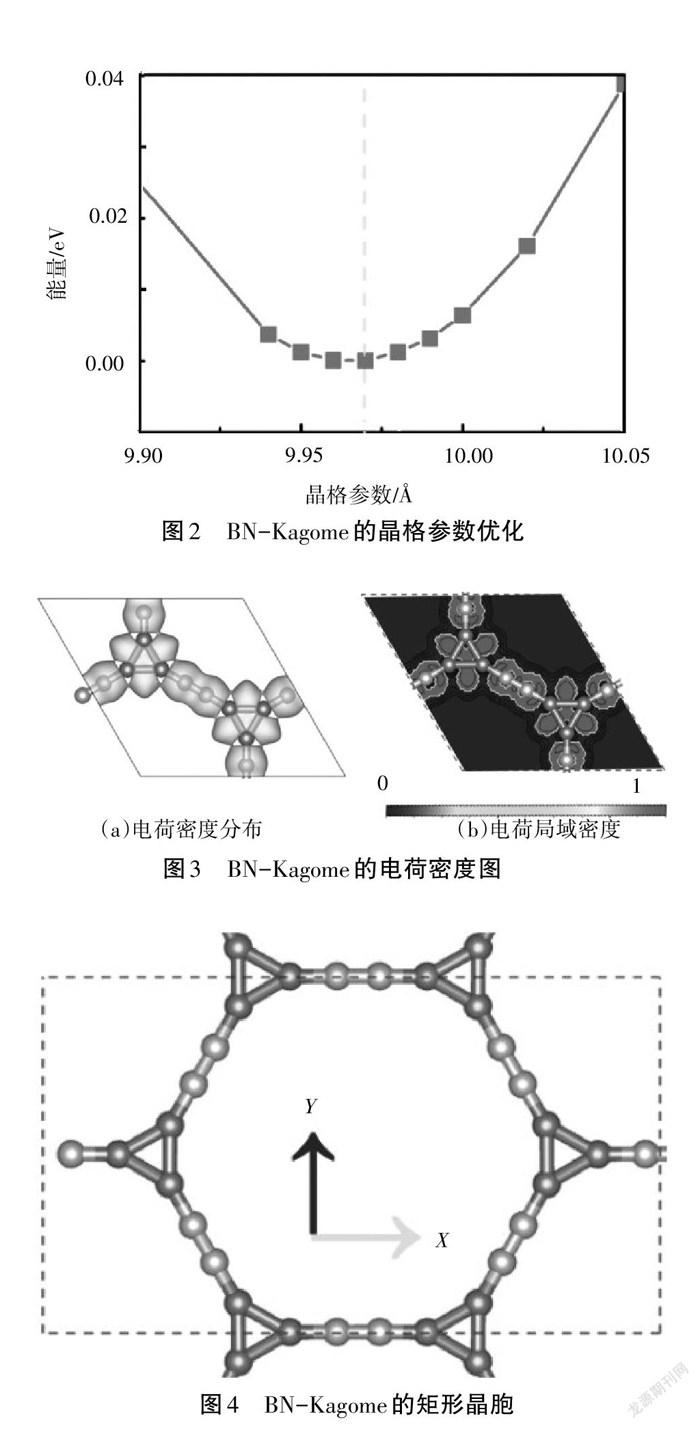
首先,建立单层BN-Kagome的原子结构,如图1所示,其为六角原胞结构,角度分别为60°和120°,原胞包含6个硼原子、6个氮原子,即化学式为B6N6[7]。然后,通过Python程序改变原胞的晶格长度构建总能量与晶格参数的关系,如图2所示。根据能量最低原理,优化的晶格参数为9.97 Å。测量B—B、B—N、N—N的键长分别为1.64 Å、1.33 Å和1.19 Å。
进一步做出了BN-Kagome的电荷分布图和电荷局域密度分布图,如图3所示。由图3可知,电荷均匀地分布在N—N和B—B键周围,而在B—N附近无电荷分布,说明N—N和B—B为共价键,而B—N为离子键。利用Bader电荷分析电子转移情况,发现B失去0.64 e,N得到0.67 e,这与电荷局域密度分布一致。
2.2 力学性质
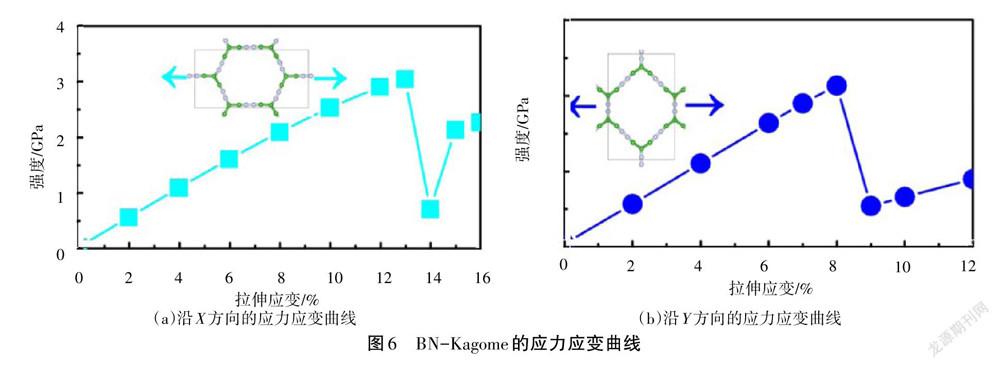
采用如图4所示的矩形超胞计算材料的弹性常数,结果为C11= 39.57 N/m、C12= 36.70 N/m、C22= 70.09 N/m 和C66= 2.10 N/m。该参数满足弹性稳定性准则C11>0、C66>0和 C11C22(2 773.46)>C12C12(1 346.89)。根据弹性常数,利用Python程序画出BN-Kagome的杨氏模量、泊松比和剪切模量,如图5所示。由图5可知,该材料表现出各向异性的力学性能。
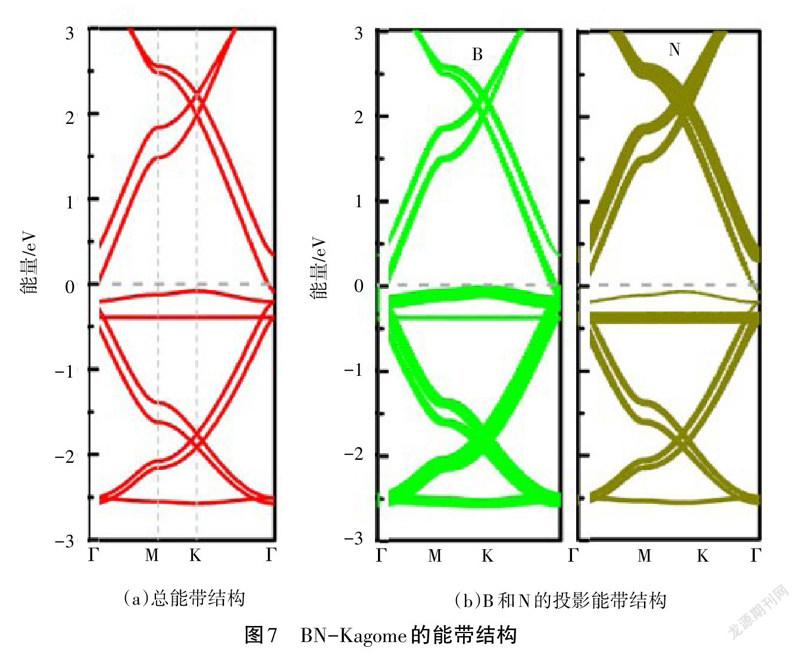
在矩形晶胞的基础上,测试沿着X方向和Y方向的拉伸应力应变曲线,如图6所示。由图6可知,沿X方向的拉伸应变为13%时断裂,强度为3.05 GPa,而沿Y方向的拉伸应变为8%时断裂,强度为2.13 GPa,这再次说明BN-Kagome矩形晶胞具有各向异性力学行为。
2.3 电子性质
采用六角原胞计算BN-Kagome的能带结构,如图7所示。图7(a)中的总能带结构显示自旋上和自旋下完全重合,表现为无磁性的基态。但费米能级附近有电子态存在,说明该材料表现为金属特性。能带结构中在-3~0 eV范围内存在两套平带和狄拉克锥,即包含两套Kagome能带。
狄拉克锥位于K点,而平带和狄拉克能带交叠于Γ点。为进一步研究能带结构特点,做出B、N的投影能带,如图7(b)所示。紧挨费米能级的平带主要由B元素贡献,而离费米能级较远的平带主要由N元素贡献,说明这两套Kagome能带来自不同的元素贡献。
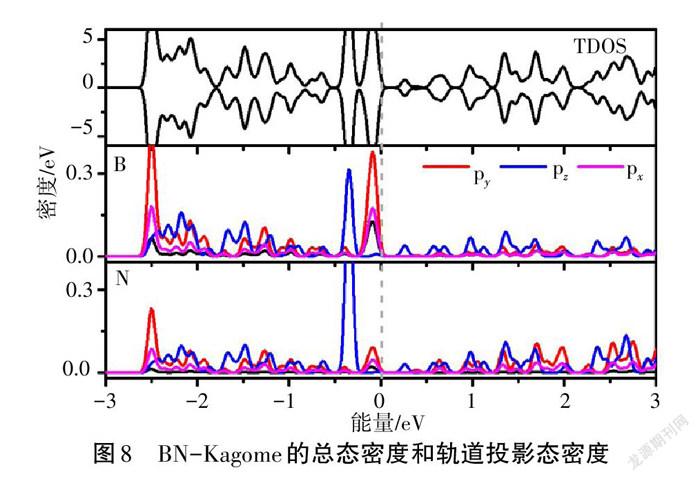
为更深入地探究Kagome能带的电子特性,做出了BN-Kagome的总态密度和B、N元素的轨道投影态密度,如图8所示。总态密度TDOS显示自旋上和自旋下的电子态分布完全对称,与能带结构一致。投影态密度显示,紧挨着费米能级的平带主要由B元素的py轨道贡献,而费米能级附近的次紧邻平带主要由N元素的pz轨道贡献。同时还可以看出,在-0.5~0 eV范围内,B-pz、B-py与N-pz、N-py发生明显交叠;在-3~-2 eV范围内,B-py与N-py交叠明显,说明二者形成牢固的化学键,这使BN-Kagome呈现出优异的结构稳定性。
2.4 对氮气分子的吸附
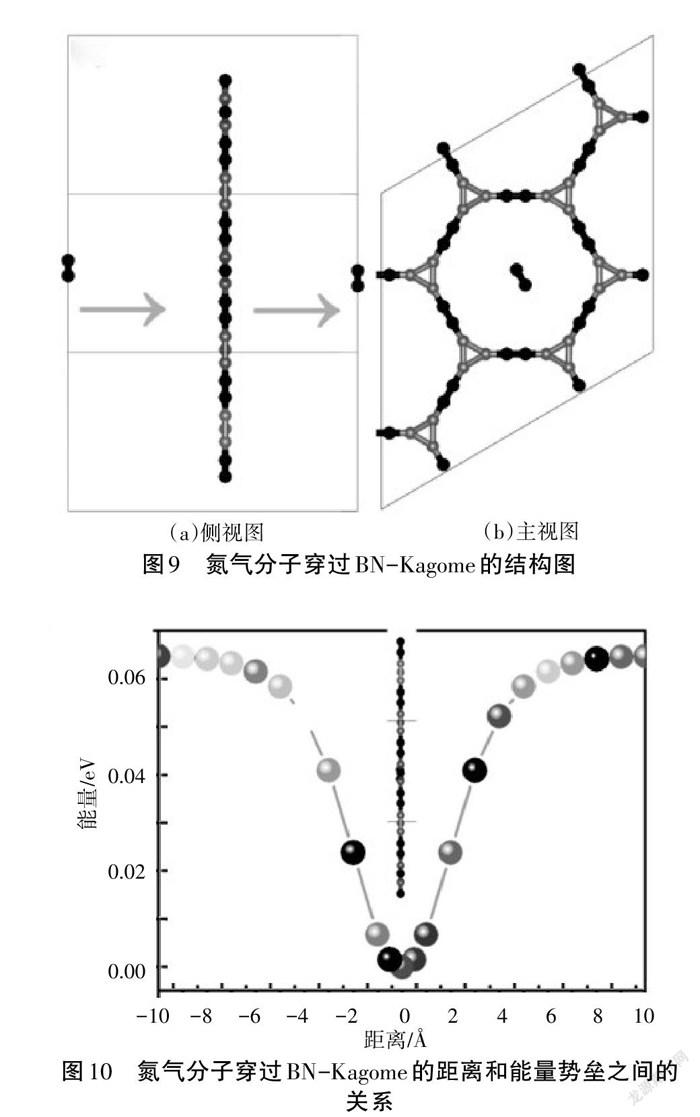
根据BN-Kagome材料的结构优化结果,可以发现该材料存在一个直径约为9.97 Å的孔洞。具有孔洞结构的材料可以用来分离过滤气体小分子,在气敏传感器件中具有广阔的应用前景。分别选择不同位点吸附氮气分子,发现氮气分子趋向于吸附在孔洞结构的中心位置,氮气分子穿过BN-Kagome的侧视图和主视图,如图9所示。接着按照图9(a)从离BN-Kagome材料的10 Å处依次按照1 Å的步长计算总能量,计算结果如图10所示。当氮气分子离BN-Kagome材料的距离大于8 Å时,总能量趋于平衡,从远离8 Å 到与BN-Kagome同一平面时总能量逐渐降低。因此,氮气分子与BN材料之间的距离为0时最稳定,该材料表现出对氮气分子的优异气敏特性。
3 结语
采用基于密度泛函理论的计算方法对二维BN-Kagome材料的原子结构、力学性质和电子结构进行了研究。BN-Kagome的原胞中包含6个硼原子和6个氮原子。N—N和B—B为共价键,而B—N为离子键。BN-Kagome材料表现出各向异性的力学性能,并表现出无磁性的金属特性。BN-Kagome材料的能带结构中存在两套Kagome能带,Kagome能带中的平带主要由B和N元素贡献。BN-Kagome材料对氮气分子吸附能较大,表现出对氮气分子的优异气敏特性。研究结果将进一步促进BN-Kagome材料的应用和发展。
参考文献:
[1] 胡凌志,商敬龙,江红民.Kagome晶格中无序和自旋交换作用对量子霍尔电导率的影响[J].宁波大学学报(理工版),2021,34:55-60.
[2] KANG M, YE L, FANG S, et al. Dirac fermions and flat bands in the ideal kagome metal FeSn [J]. Nature Materials, 2020(2): 163-169.
[3] LIU Z, LI M, WANG Q, et al. Orbital-selective Dirac fermions and extremely flat bands in frustrated kagome-lattice metal CoSn [J]. Nature Communications,2020(1): 4002.
[4] KRESSE G, FURTHMULLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Computational Materials Sciences, 1996(1): 15-50.
[5] KRESSE G, FURTHMULLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Physical Review B, 1996(16): 11169-11186.
[6] WANG V, XU N, LIU J C, et al. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code [J]. Computer Physics Communications, 2021, 267: 108033.
[7] 康玉嬌.二维氮化物薄膜材料的电子性质[D].湘潭:湘潭大学,2020.
