神经嵴细胞和神经嵴病及其致病机制的研究进展
2022-03-08蒋卓远查艳石小峰张永彪2
蒋卓远,查艳,石小峰,张永彪2,
综 述
神经嵴细胞和神经嵴病及其致病机制的研究进展
蒋卓远1,查艳1,石小峰3,4,张永彪2,3,4
1. 北京航空航天大学生物与医学工程学院,北京 100000 2. 北京航空航天大学医学科学与工程学院,北京 100000 3. 北京航空航天大学大数据精准医疗高精尖创新中心,北京 100000 4. 工信部大数据精准医疗重点实验室,北京 100000
神经嵴细胞(neural crest cells,NCCs)是一类脊椎动物特有的可迁移的多能干细胞,其可分化为软骨细胞、神经元和黑色素细胞等多种类型细胞。NCCs的形成、迁移和分化受到严格调控,任何扰乱NCCs发育的因素都可导致胚胎发育畸形。由神经嵴细胞发育异常所导致的一系列疾病统称为神经嵴病(neurocristopathies,NCPs)。NCPs种类繁多且表型复杂,可累及人体多个部位(颅面部、心脏、肠胃和皮肤等),严重危害患者的身体机能和心理健康。NCPs占所有出生缺陷患儿的1/3,遗传因素是导致NCPs的主要风险因素,但环境风险因子以及基因–环境交互作用异常也可导致NCPs。本文对神经嵴细胞和神经嵴病及其致病机制进行综述,为系统认知神经嵴细胞发育以及神经嵴病提供参考,为了解神经嵴病的病因以及开展有效防控提供科学支撑。
神经嵴细胞;神经嵴病;基因调控网络;风险因素;发病机制
神经嵴细胞(neural crest cells,NCCs)是脊椎动物胚胎发育早期的一种具备全胚胎迁移性的全能性干细胞[1]。NCCs经过了从躯干向头部逐步特化的过程,对脊椎动物头颅的演化起到至关重要的作用[2,3]。NCCs赋予了脊椎动物高级的感官系统、复杂且精细的神经系统以及强有力的掠食器官,支持脊椎动物从被动的滤食行为演化到主动捕食的生活方式[4]。NCCs起源于背侧外胚层的神经板边界,神经褶皱的融合诱导了神经管的闭合以及NCCs的形成。随后,神经嵴细胞会有序地从胚胎背侧向腹侧迁移,在迁移过程中以二元命运决定方式分化成为祖细胞和间质细胞,进而继续二元命运决定,分化为软骨细胞、色素细胞、平滑肌细胞和神经元等[5,6]。除此之外,迁移的NCCs还在胚胎发育早期替代未发育的吞噬类细胞发挥死细胞的清除功能[7]。NCCs虽然来自外胚层,但其仍具备分化为中胚层和内胚层的能力,因此,有学者将神经嵴称为“第四胚层”,甚至其多能性特征高于其他三个胚层[8]。
NCCs赋予脊椎动物演化优势的同时,也引入了发育畸形风险。NCCs的发育过程始终受基因严格调控,任何异常调控都将扰乱NCCs正常发育,进而导致胚胎发育畸形。1974年,Bolande等[9]统梳理了NCCs衍生物异常发育的系列疾病,并统称为神经嵴病(neurocristopathies,NCPs)。已知的颅面部发育缺陷大多数和神经嵴细胞发育相关,而颅面部出生缺陷患者几乎占所有人类出生缺陷的1/3[10],他们临床症状复杂,病因大多未知,分类较为混乱。导致该现状的主要原因有:(1)NCPs种类繁多,多为散发罕见病例,样本收集困难,导致难以进行系统研究;(2)不同NCPs临床特征纷乱交叠,NCPs的病例症状多样、表型冗杂交叠,目前未形成系统有效的分类体系;(3)NCPs病因缺乏系统揭示,NCPs基本都是独立研究,如典型代表——唇腭裂和先天性巨结肠(Hirschsprung),但在NCCs层面系统的研究各类NCPs的工作仍未开展[11]。本文依据NCPs的受累器官对其进行系统分类,然后以NCCs的发育为线索,对NCPs的病因进行梳理,总结了NCCs发育异常导致各类NCPs的机制。
1 神经嵴细胞
1.1 NCCs的形成
NCCs来源于神经板边界(neural plate border,NPB)内的神经嵴祖细胞(图1)。在人类早期胚胎发育过程中,神经嵴祖细胞可诱导促使神经板两侧的NPB隆起,形成神经褶皱,随后隆起的神经褶皱融合形成神经管,神经嵴祖细胞聚集于神经管背侧区域,以等待调控因子提供特化(specification)信号[1]。
NCCs的形成分为诱导(induction)、特化(specification)和预迁移(pre-migration)三个阶段(图1)。小鼠模型研究揭示NCCs形成过程由多个调控通路参与,包括骨形态发生蛋白(Bmp)信号通路、Wnt信号通路、成纤维细胞生长因子(Fgf)信号通路和Notch信号通路[1]。研究发现,Bmp信号在NCCs诱导早期开始发挥作用,然后激活Wnt信号在NCCs的特化阶段发挥调控活性,他们均对该阶段NCCs数量的维持起关键作用[12]。Fgf信号主要在诱导过程中发挥作用,Fgf8信号的中断阻止了NPB基因的表达[13]。
1.2 NCCs的迁移
NCCs的迁移始于神经管闭合,迁移之初,NCCs经上皮–间充质转化(epithelial-mesenchymal transition,EMT)过程从神经管背侧剥离,然后开始迁移。NCCs的EMT过程是从形成态向迁移态的转换,转换后的细胞命运由一系列调控因子决定。Soldatov等[6]发现在NCCs的命运决定在迁移时就已发生,NCCs迁移前阶段就分化为两个不同的亚群:第一是最早的EMT前群,它们由未分层(delamination)的NCCs组成;第二是分层的细胞亚群,其特征在于EMT的关键基因的激活和的沉默,该亚群细胞还被一系列基因的连续瞬时调控,如、、和。之后细胞命运决定具体表现为一系列连续的二元甚至多元决定过程,该过程首先表现为NCCs向感觉类的神经胶质细胞和其他过渡类型细胞的身份转换,随后过渡类型细胞再向自主神经细胞和间质细胞身份转化的二元命运决定过程,这两类细胞继续后续的命运决定过程,直至分化为最终细胞类型。
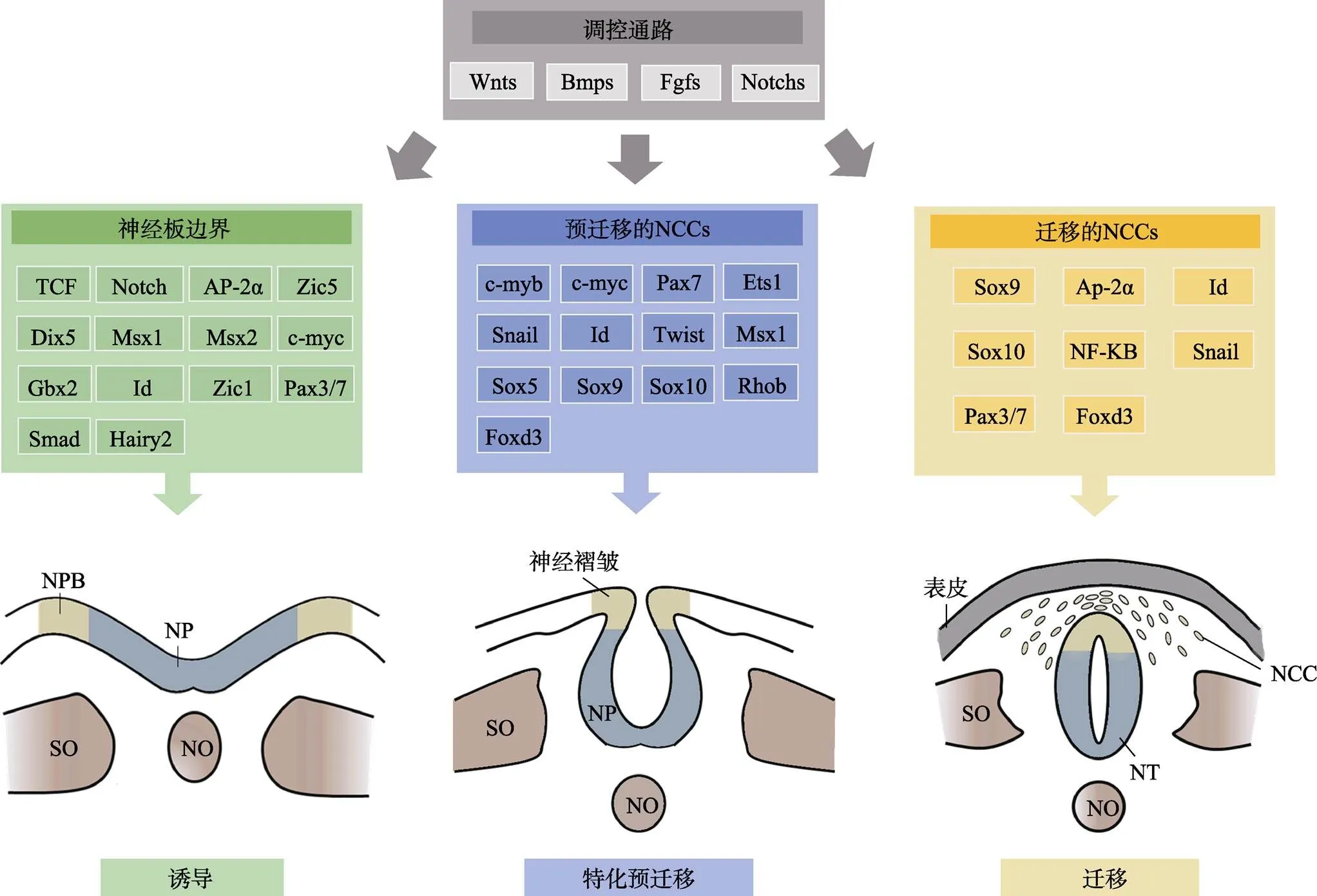
图1 NCCs的发育过程受信号因子的严格调控
NCCs的形成始于神经板边界,主要由Wnts、Bmps、Fgfs和Notchs信号调节。在神经管形成早期,神经嵴祖细胞位于NPB中,其诱导神经板(neural plate,NP)两侧的NPB隆起形成神经褶皱,两侧的神经褶皱融合形成神经管后,NCCs便开始诱导–特化–预迁移过程。在大多数脊椎动物中,NCCs的分层及迁移始于神经管闭合,但该过程也可早于神经管闭合,比如小鼠的NCCs分层在神经管未闭合时即发生。NCCs:神经嵴细胞;NPB:神经板边界;NF:神经褶皱;SO:体节;NT:神经管;NO:脊索。
根据NCCs的形成位置和迁移路线,可将其分为四种类型:颅、躯干、迷走和骶NCCs,迁移后在胚胎的各部位分化成多种功能细胞(图2:A和B)。颅NCCs从神经管背侧迁移出来后,将分裂成三束分散(discrete streams)的细胞流:迁移至中、前脑的背侧细胞流,迁移入鳃弓的腹侧细胞流以及从后脑向其他部位迁移的细胞流[14]。躯干NCCs从第7~28体节的神经管向外迁移的路线有两条:一条途径沿背外侧呈连续状迁移(continuous wave),主要发育为色素细胞;另一条途径沿腹内侧呈分段式(segmented manner)迁移,主要发育为胶质和神经元前体[15]。迷走NCCs从菱形脑节4~7产生后主要迁移到心脏,同时也像骶NCCs迁移到肠道定植,这个是NCCs最长的一条迁移路线[16]。神经嵴细胞的迁移受到了多种信号的严格调控[17](图2C),包括局部抑制剂,诱导剂和细胞粘附分子,如Snails可直接抑制细胞粘附分子E-cadherin,促进细胞迁移,Eph/ephrin、内皮素和Slit/Robo信号决定了NCCs的背外侧和腹内侧迁移路径的选择。
最新研究发现NCCs的迁移行为还和外周环境存在交互,该过程赋予NCCs在早期胚胎发育中发挥特殊的功能:在特异吞噬细胞出现之前,迁移的NCCs被证实能在神经系统中承担吞噬细胞碎片的功能。该吞噬过程与巨噬细胞吞噬相似,均由白细胞介素-1所介导[7]。现阶段,NCCs在迁移过程中的时空调控网络以及与周围细胞的互作关系还亟待深入研究,明晰其迁移转折点的决策机制也将成为后续研究热点。
1.3 NCCs的分化
NCCs最终分化成何种细胞和解剖结构类型,主要取决于终点微环境。当NCCs遇到血管或细胞簇
等提供的终止迁移信号时,便开始分化[11]。颅神经嵴细胞(cranial neural crest cells,CNCCs)最终迁移至头部和咽弓,参与颅面部的骨骼,皮肤,神经和肌肉的形成。其他部位的NCCs最终分化三个为:迷走神经嵴细胞参与主动脉肺隔、心神经节和肠神经节的形成;躯干神经嵴细胞发育为黑色素细胞、神经元、神经胶质和施旺细胞;骶神经嵴细胞分化成交感神经节和肠神经节[11,18](图2:B和D)。
2 神经嵴病
2.1 神经嵴病的分类
1974年,Bolande等[9]将由NCCs及其衍生物异常发育引起的一系列疾病统称为NCPs,如Treacher Collin综合征、唇腭裂、Waardenburg综合征、先天性巨结肠。随着研究的深入,那些由于核糖体或纤毛等具体结构发育异常进而导致NCCs发育异常的疾病也被列入NCPs范畴,如核糖体病、纤毛病等。然而由于神经嵴细胞参与了机体大量细胞和器官的发育,从而导致神经嵴病数量众多,加上NCPs表型多样以及病因认知不足导致神经嵴病未被系统梳理。1977年,Reed等[19]依照是否伴发肿瘤将NCPs划分为发育异常(dysplasias)、肿瘤(neoplasms)以及畸形和肿瘤并存的三种类型。2018年,Vega-Lopea等[11]按照神经嵴细胞的来源将神经嵴病分为颅神经嵴病、躯干神经嵴病、迷走神经嵴病和骶神经嵴病。但上述分类方法过于简单,不利于对NCPs的系统认知。本文将按照神经嵴病的主要累及器官,对已知的神经嵴病进行分类,同时将和NCCs相关的肿瘤单独举出为一类(图3)。
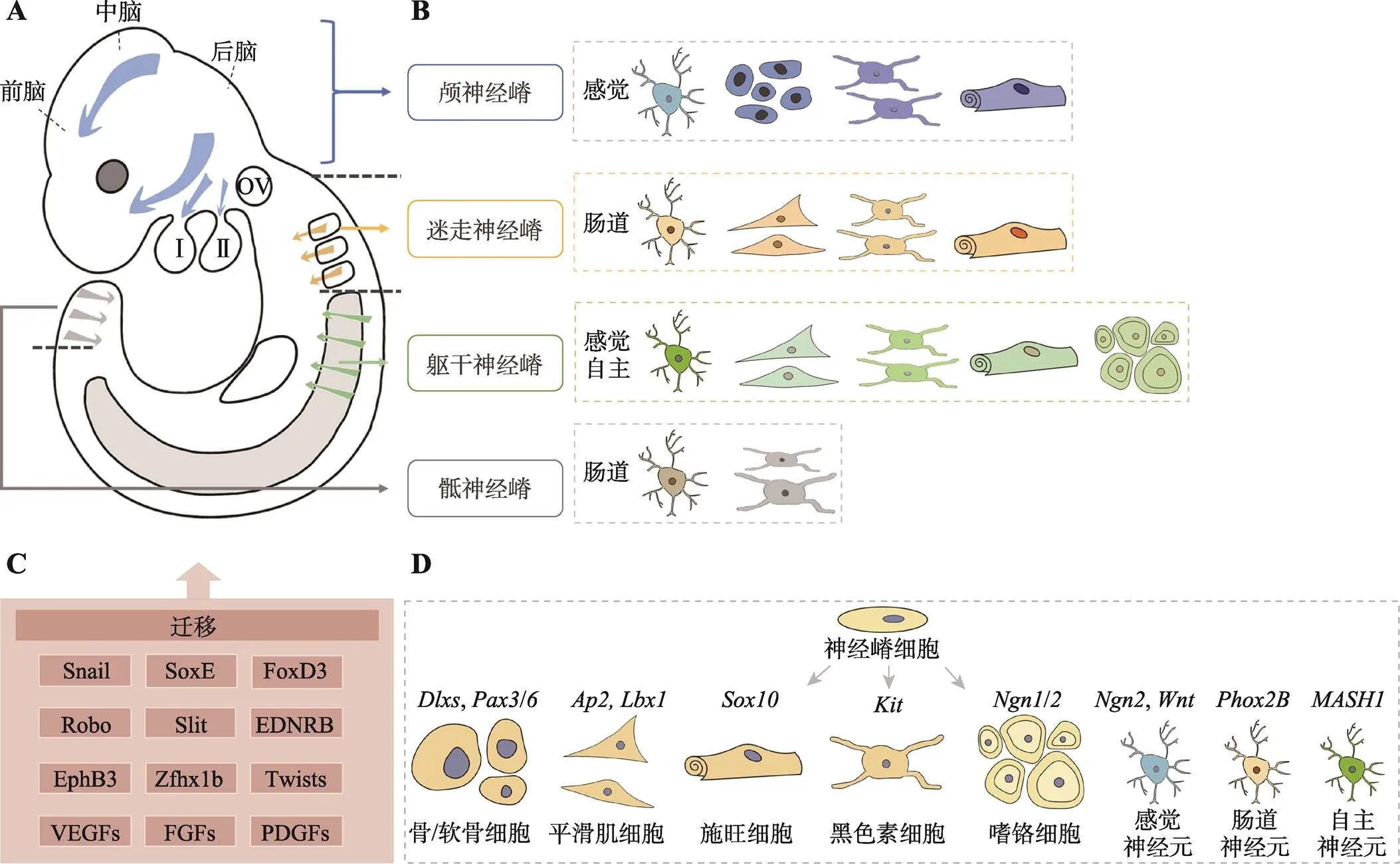
图2 NCCs的迁移和分化
A:NCCs在胚胎中的迁移路径。NCCs分为颅神经嵴、迷走神经嵴、躯干神经嵴和骶神经嵴四个类型。箭头表示NCCs的迁移流。B:NCCs可分化的组成机体的主要细胞类型。C:NCCs迁移过程的调控因子和信号通路。D:调控NCCs分化为各种细胞类型的关键调控信号。Ⅰ、Ⅱ:第一/二咽弓;OV:听囊。
2.2 神经嵴病的典型病症
神经嵴病可发生在全身多个部位,或累及单个器官,或同时累及多个器官,形成复杂的神经嵴疾病。本文根据人体的解剖结构进行划分,总结了NCPs的典型表型,列举了NCPs引起的颅面、脏器、四肢、皮肤及全身发育的异常[20](表1)。颅面部畸形是最常见的典型特征,通常包括上、下颌不完全发育,牙齿形态异常,进食和说话困难。下颌的严重发育不良会使婴幼儿的气管受到压迫,气管狭窄,从而影响呼吸,甚至导致死亡。部分患者眼睛歪斜、睫毛稀疏、眼睛色素异常,导致视力下降。耳朵发育异常也是多种NCPs的典型特征,如外耳发育不全或缺失(小耳或无耳)、耳道闭锁、听力丧失等[21]。
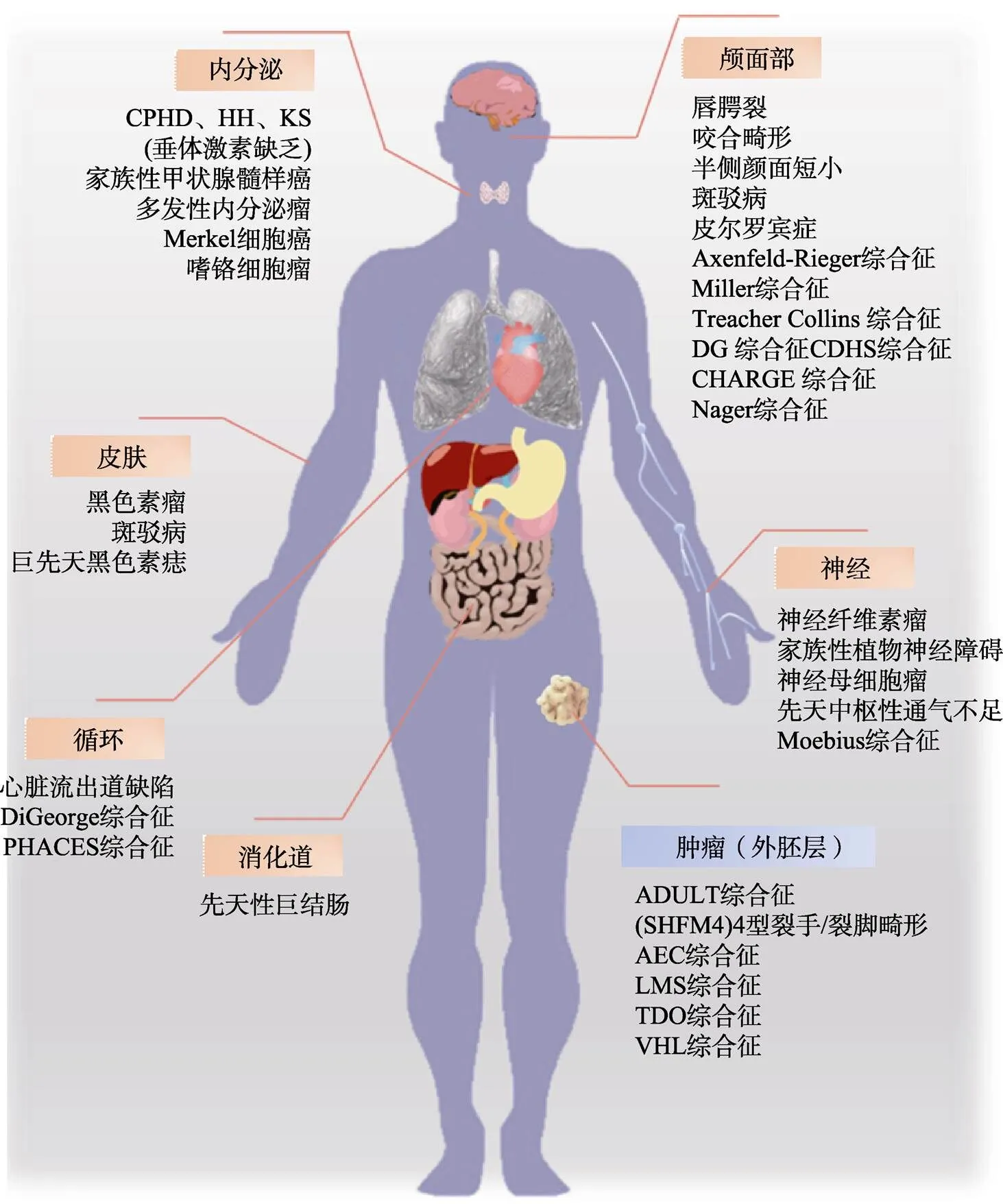
图3 NCCs发育异常导致的神经嵴病所累及的人体器官及系统
按照神经嵴病的主要累及器官,对已知的神经嵴病进行分类。如唇腭裂、半侧颜面短小等都是具有典型颅面部症状的神经嵴病,先天性巨结肠是典型的消化道NCP。CDHS:颅面–耳聋–手综合征(craniofacial-deafness-hand syndrome);SHFM4:裂手/裂脚畸形4型(split-hand/split-foot malformation type 4);LMS:四肢-乳腺综合征(limb-mammary syndrome);TDO:毛齿骨综合征(tricho-dento-osseous syndrome);VHL:Von Hippel-Lindau综合征(Von Hippel-Lindau syndrome)。
2.3 神经嵴病流行病学分析
神经嵴病占所有先天性出生缺陷患儿的1/3[10]。据统计,颌面部第一高发出生缺陷——唇腭裂发病率为1/500~1/1000[27],而颌面部受累器官最多的半侧颜面短小畸形的发生率在1/5600~1/6550[22]。1岁以下婴儿最常见的癌症——神经母细胞瘤发病率为每1/10万新生儿。消化道典型的神经嵴病——先天性巨结肠(Hirschsprung disease,HSCR)的发病率约为1/5000新生儿[11]。然而,神经嵴病的发病率很可能被低估了,原因在于医生对NCPs的本身及其变异性的认识不足,轻微表型畸形得不到重视,以及疾病无法得到充分且精确诊断。NCPs发病部位的一个典型特征是存在机体的不对称性,表现为大部分受累器官仅出现在单侧(表2)。
2.4 神经嵴病的防治
当前对于遗传疾病,以防控为主,诊疗为辅。由于大量神经嵴病患者颌面部畸形明显,不仅给患者造成身心伤害,后期的矫正性治疗还将给家庭和社会带来巨大经济压力。将防控关口前移是出生缺陷防治的重要方向,对存在遗传风险家族史的孕妇采取产前检测可有效避免畸形胎儿出生。而在孕期进行适当的防控管理,如补充叶酸,避免暴露在致畸剂,如酒精、尼古丁、维甲酸、甲醛等是NCPs防控的较优策略。

表1 神经嵴病发病部位和临床表型
TCS:Treacher Collin综合征(Treacher Collins syndrome);CS:颅缝早闭(craniosynostosis);HFM:半侧颜面短小畸形(hemifacial microsomia);DBA:先天性再生障碍性贫血(diamond blackfan anemia);CADSIL:大脑常染色体显性动脉病变伴皮层下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy);PHACES:后窝畸形–血管瘤–动脉异常–心脏缺陷–眼睛异常–胸骨裂和脐上中缝(posterior fossa malformations–hemangiomas–arterial anomalies–cardiac defects–eye abnormalities–sternal cleft and supraumbilical raphe);HSCR:先天巨结肠(Hirschsprung disease);AAA:三联A综合征(achalasia- addisonianism-alacrima):CCHS:先天性中枢性通气不足症候群(congenital central hypoventilation);NAS:Nager 综合征(nager syndrome);NS:努南综合征(noonan syndrome);OCA:眼–皮肤白化病(oculocutaneous albinism);HED:少汗性外胚层发育不良(hypohidrotic ectodermal dysplasia;CPHD:垂体激素缺乏症(combined pituitary hormone deficiency);GDFD:发育迟缓,面部畸形(growth retardation, developmental delay, and facial dysmorphism)。
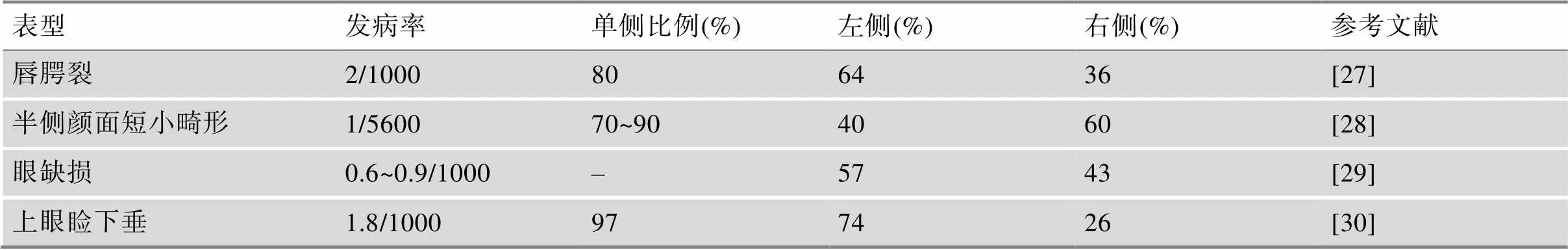
表2 代表性神经嵴病的发生率及患侧发生比例
体表畸形的神经嵴病的治疗主要运用手术矫正,如唇腭裂、小耳畸形等。NCPs相关肿瘤的治疗也已实现靶向药物治疗和低剂量化疗,如神经母细胞瘤。体外协助治疗也是行之有效的手段,对于听力丧失和呼吸困难的患者采取助听器和通气治疗,可极大改善患者生活质量甚至挽救患者生命。随着科技的不断进步,基因治疗和干细胞治疗未来可期:干预基因进而矫正NCCs在TCS患者中的异常发育已经在小鼠模型上获得成功[31];母体抗氧化剂修复DNA损伤防止NCCs细胞凋亡可能成为预防神经嵴病的有效手段[32];利用干细胞再生技术实现受损或畸形的组织修复将是未来NCPs治疗研究的重心。
3 神经嵴病的病因学
众多转录因子和信号分子协同调控了神经嵴细胞的复杂发育过程。在该过程中,任何调控异常都会扰乱NCCs的形成、迁移和分化,导致NCPs的产生。现在公认的神经嵴病的致病因素是:NCCs基因调控网络(genetic regulation network,GRN)内的基因突变、表观修饰异常以及环境致畸因素。由于NCPs种类众多,GRN调控信号复杂,导致GRN的基因突变、NCCs发育过程和神经嵴病三者的关系难以系统总结。下面根据NCCs发育过程中基因发挥调控活性的时序(附图1),梳理了神经嵴病的发病机制。
3.1 基因调控网络异常
3.1.1 NCCs形成异常所致的神经嵴病
NCCs的形成是一个多步骤的过程。在NCCs形成前后,、和等神经板边界调控基因的开始表达,紧随其后的是更具特异性的NCCs调控基因,包括、和等[33,34]。其中作为转录因子的SOX9、PAX3、TFAP2A、FOXD3等信号异常,会影响NCCs的形成过程,导致细胞分裂和细胞的凋亡异常(表3)。在神经嵴细胞的生长和增殖过程中,核糖体扮演着重要的角色。核糖体会将信使RNA(mRNA)翻译成氨基酸序列,参与细胞内蛋白的合成。本文以NCCs形成异常所导致的典型神经嵴病——核糖体病为例,阐述其发病机制。
核糖体的形成过程如下:在多种转录因子的调控下,首先核糖体DNA(rDNA)会被转录成pre- rRNA。pre-rRNA生成后,会生成一个中间过渡的转录本,然后再被加工成成熟的rRNA。RPs与rRNA组装成核糖体的小亚基(40S)和大亚基(60S),最后会被运输到细胞质中进一步形成成熟的核糖体。该过程中,某些基因的突变会造成核糖体的合成异常导致NCPs,其致病机制是因核糖体的合成不足,最终导致NCCs分裂异常或细胞凋亡程序失控,导致NCCs形成不足[35]。由于相关基因突变导致核糖体合成异常而影响NCCs形成所产生的NCPs,称为核糖体病,它是NCPs的一个重要的亚型[36](图4)。
3.1.2 NCCs迁移异常所致的神经嵴病
神经嵴细胞的迁移过程由细胞内的自主信号以及在迁移过程中与细胞相互作用的周围环境共同协调调控[14,17,56,57]。在分层阶段,Snails转录因子家族发挥了主要的作用。、等基因也是NCCs迁移所必需的,其突变被证实会影响哺乳动物NCCs的分层和迁移。斑驳病、家族性自主神经障碍(FD)和II型瓦登伯格综合征(WSⅡ)等都是典型的由于NCCs迁移异常所导致的神经嵴病(表3)[31~33]。
迷走和骶NCCs会迁移到肠道,在肠道形成肠道神经元,组成肠道神经系统(enteric nervous system,ENS)。许多营养因子、细胞表面受体、转录因子和信号分子参与了此迁移活动,如Eph/Ephrin、BMP、RET信号等。其中RET是一种跨膜蛋白,受到BMP信号调控,参与ENS的存活,增殖,迁移和分化过程。EDNRB与RET信号协同激活MAKP和PI3K通路下游信号和调控基因,如和基因突变会影响ENS的发育,导致先天性巨结肠症。先天性巨结肠的主要病因是由于ENS前体细胞不能定植胎儿肠远端,导致肠道神经元缺失,出现肠梗阻(图5)。
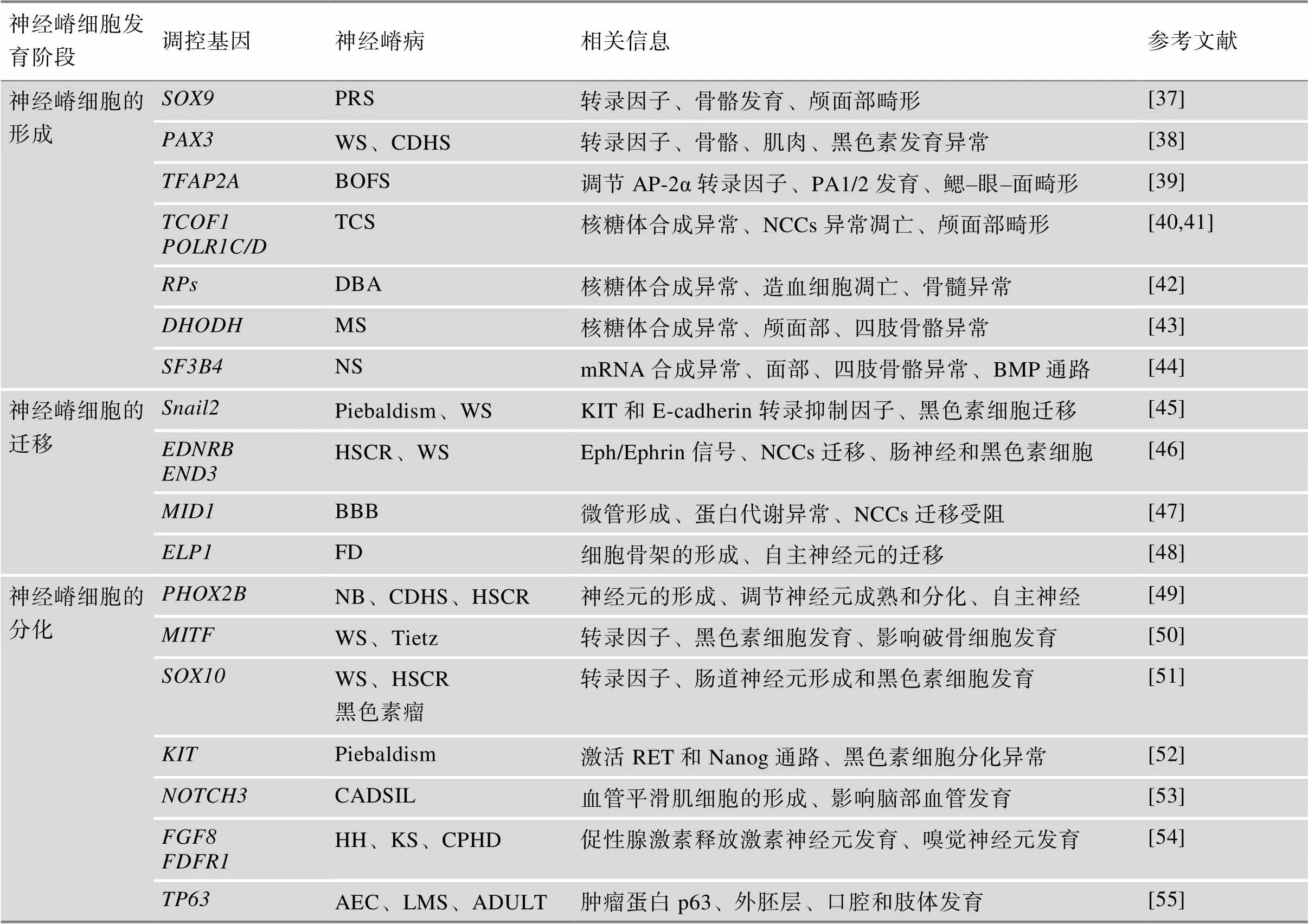
表3 基因调控网络异常导致的神经嵴病
PA1/2:第1/2鳃弓;PRS:皮埃尔·罗宾序列(Pierre Robin sequence);WS:瓦登伯格综合征(Waardenburg syndrome);CDHS:颅面–耳聋–手综合征(craniofacial-deafness-hand syndrome);BOFS:鳃–眼–面部综合征(branchio-oculo-facial syndrome);TCS:Treacher Collins综合征(Treacher Collins syndrome);DBA:先天性再生障碍性贫血(diamond blackfan anemia);MS:米勒综合征(Miller syndrome);NS:努南综合征(noonan syndrome);HSCR:先天性巨结肠症(Hirschsprung disease);BBB:Opitz G/BBB综合征(opitz g/bbb syndrome);FD:家族性自主神经障碍(familial dysautonomia);NB:成神经细胞瘤(neuroblastoma);Tietz:Tietz综合征(Tietz syndrome);CADSIL:常染色体显性遗传病合并皮质下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy);HH:性腺功能减退症(hypogonadotropic hypogonadism);KS:Kallmann综合征(Kallmann Syndrome);CPHD:合并垂体激素缺乏(combined pituitary hormone deficiency);AEC:AEC综合征(aec syndrome);LMS:四肢–乳腺综合征(limb-mammary syndrome)。
3.1.3 NCCs分化异常所致的神经嵴病
神经嵴细胞的分化是细胞命运决定的结果,一些转录因子定义了NCCs的初始分裂状态,并赋予了它向不同的谱系分化的潜力。在NCCs迁移过程中,和就已经参与了NCCs的命运决定:在NCCs发育过程的初期开始表达,它决定了NCCs向间充质细胞(骨、软骨和结缔组织)分化的潜能,而则定义了早期的感觉神经NCCs祖细胞,将它们和色素NCCs祖细胞进行区分[6]。之后再通过各类特异性因子调控,NCCs得以分化形成不同类型的细胞,如PHOX2B与神经元的形成相关、MITF与黑色素细胞发育相关、FGF9与嗅神经发育相关和NOTCH3与血管平滑肌细胞相关(表3)。
黑色素细胞是NCCs的重要衍生细胞之一,参与了皮肤色素、视网膜色素的形成,还参与内耳的发育,影响胎儿听力。编码黑素细胞诱导转录因子MITF,控制黑色素细胞发育及其功能。MITF还受到其他NCCs特异基因的调控:PAX3与SOX10能反式激活其启动子,协同上调的表达[58]。肠道神经元分化的调控信号和黑色素细胞的调控网络存在交互作用,、和除了在NCCs来源的黑色素细胞表达外,也是肠神经元细胞增殖、迁移和生存的关键基因。以上几种基因中的任何突变都会导致皮肤黑色素细胞和肠道中神经元的缺失,引起WS和斑驳病(图6)。
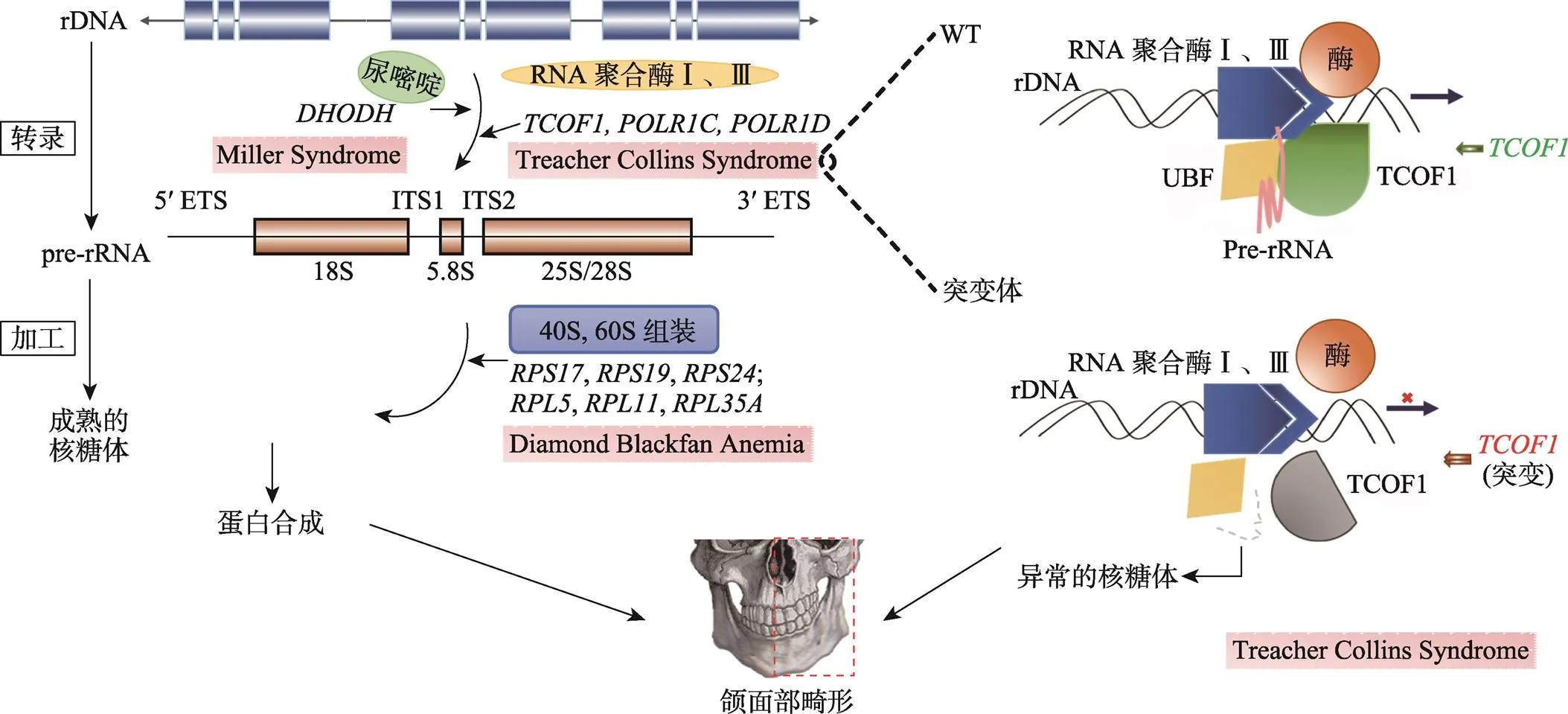
图4 核糖体的合成异常导致的神经嵴病的发病机制
与NCPs相关的基因突变会导致核糖体的合成异常,造成颅面部畸形。RNA聚合酶I和III参与rDNA转录,Treacher Collin综合征中、和的突变会破坏转录复合物的形成,影响rRNA的产生。尿嘧啶也参与rRNA的合成,Miller综合征中的突变干扰尿嘧啶的合成。在RNA聚合酶I和III合成pre-rRNA后,基因(、等)突变破坏核糖体的合成,导致DBA。rDNA:核糖体DNA;pre-rRNA:前核糖体RNA。
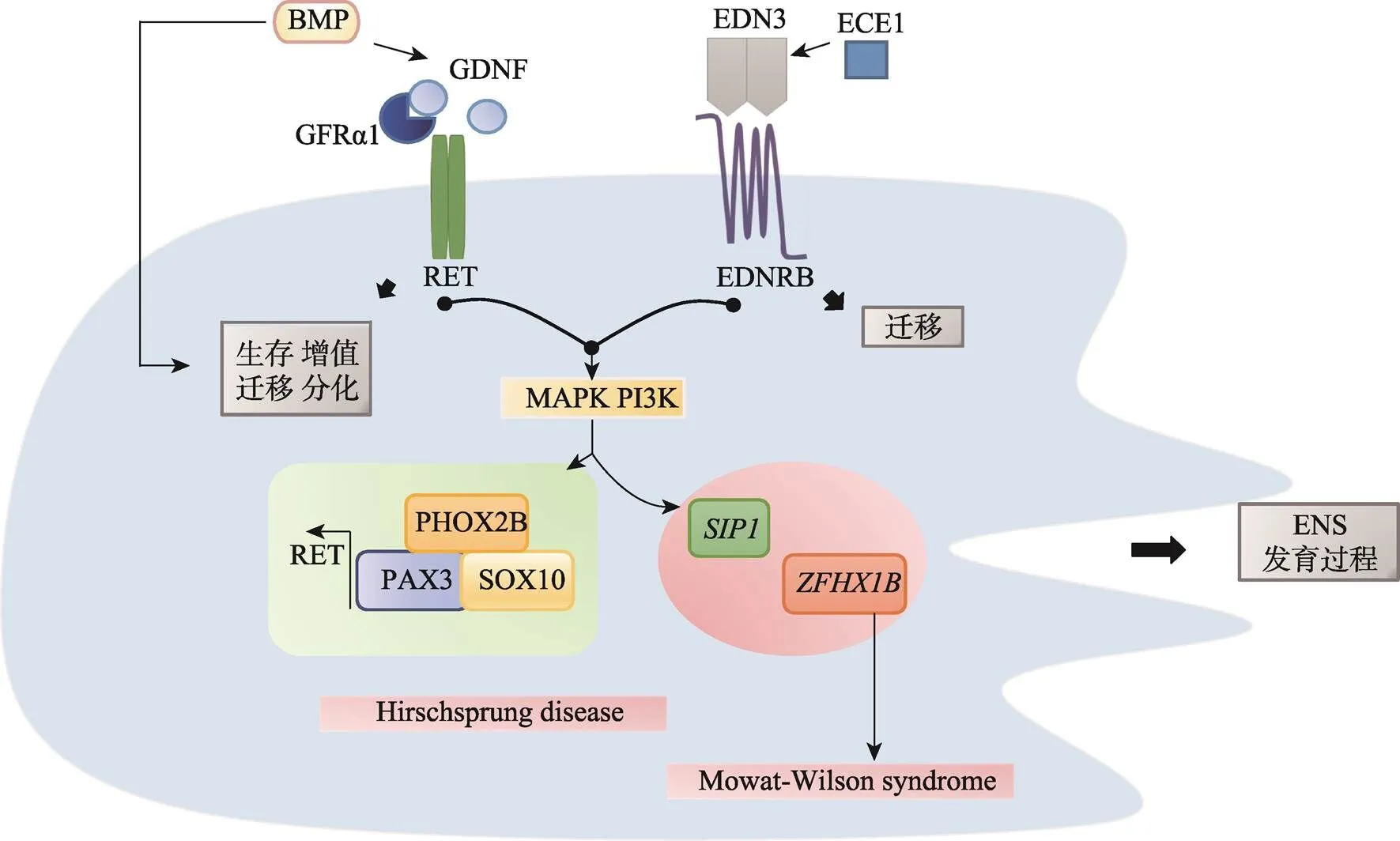
图5 周围神经细胞异常神经嵴病的发病机制
RET是一种跨膜酪氨酸蛋白,RET信号功能涉及ENS的存活、增殖、迁移和分化。RET也受到BMP信号调控,影响ENS的迁移。RET和EDNRB激活MAKP PI3K通路下游信号:与肠神经发育相关的PAX3、SOX10和PHOX2B协同激活RET转录。和突变也将影响ENS的发育,导致先天性巨结肠。Hirschsprung disease:先天巨结肠症;Mowat-Wilson syndrome:Mowat-Wilson综合征。
3.2 表观遗传因素
表观遗传学即DNA和RNA等大分子的修饰,在NCCs的发育中也起着重要的调控作用。表观遗传机制是微调基因表达和细胞分化的关键因素,它将影响基因表达并传递给后代。探索NCCs的表观遗传学机制,有助于了解NCPs的病因[59]。根据表观修饰类型的不同,表4总结了涉及突变影响的修饰基团、靶基因以及NCCs发育阶段,纳入了最新发现的microRNA的调控异常导致的NCPs,梳理了不同表观遗传修饰异常所导致的神经嵴病。
3.3 环境因素
神经嵴病的病因可归于内在遗传因素和外在环境因素的异常导致胚胎发育畸形[80]。研究发现孕妇能通过食物、饮用水、药物和空气传播以及皮肤接触到有害的环境因素,这些都将影响NCCs的发育过程。据估计,大约10%~15%的先天出生类缺陷是由于环境因素(致畸剂)对胚胎发育的不利影响所导致的[81]。另外,环境危险因素还可能通过与遗传因素的相互作用影响NCCs的发育,如Xu等[22]通过基因–环境交互作用检验研究半侧颜面短小(hemifacial microsomia,HFM)风险因素时,发现妊娠期父亲吸烟与HFM相关SNP(单核苷酸多态性,single nucleotide polymorphism)存在显著交互作用,提示吸烟暴露会增加CFM风险。另外大量的分子、药物和致畸剂或胎儿毒性物质也会影响NCCs的发育:高糖、维甲酸、四氢大麻酚、可卡因、吗啡、氯胺酮、血管紧张素转换酶抑制剂、波生坦、氯霉素、卡马西平、氟康唑、草甘膦除草剂、锂化合物、甲氨蝶呤、苯巴比妥、他莫昔芬、沙利度胺、甲苯、曲妥珠单抗、维甲酸、丙戊酸等[81]。下面简要描述一些典型的导致NCCs发育异常的环境因素的致畸源作为参考。
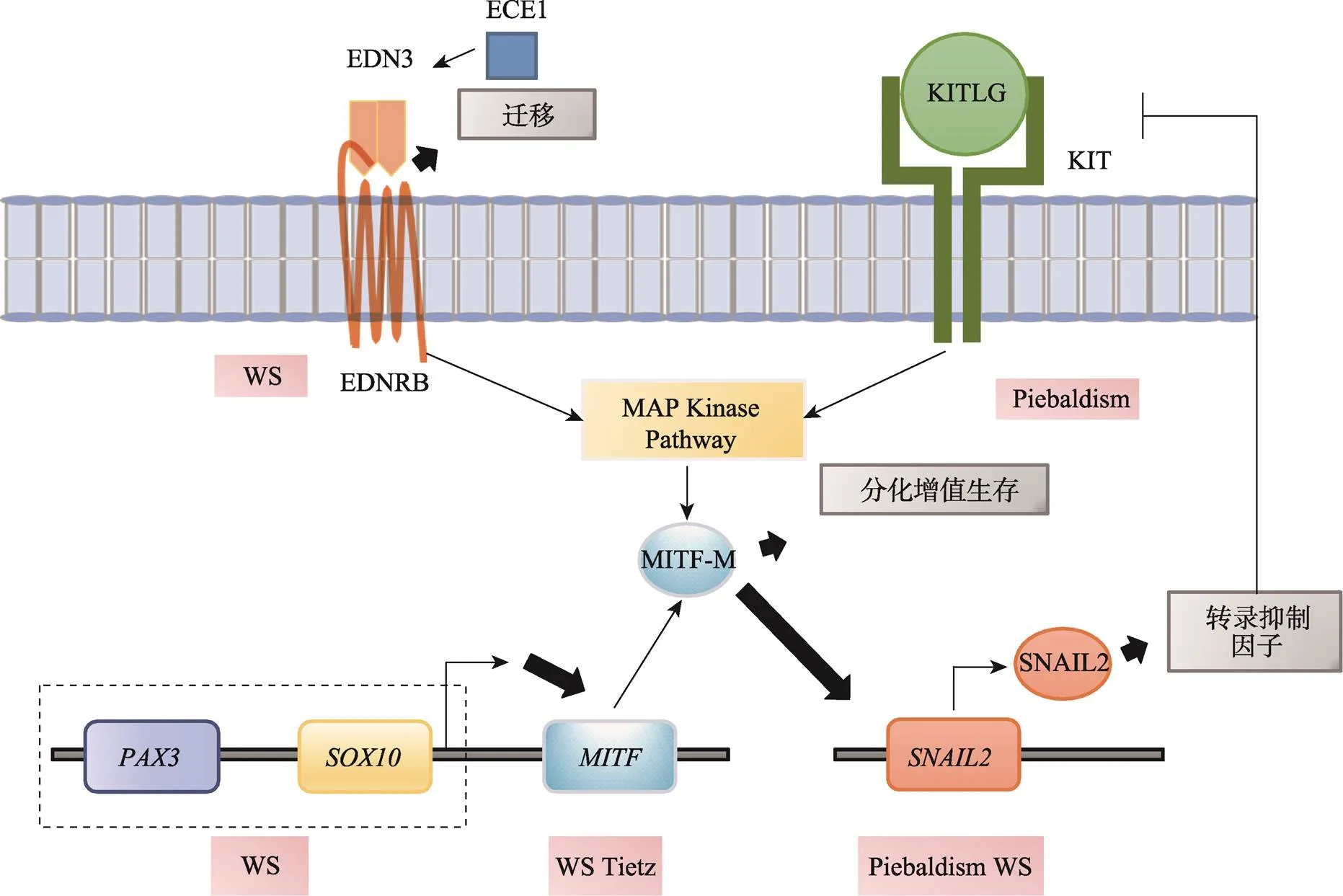
图6 黑色素细胞发育异常神经嵴病
EDNRB和KIT传导的信号激活MAPK通路,之后作用于MITF的M亚型,该亚型只在黑色素细胞谱系中表达。控制黑色素细胞的发育和细胞功能。PAX3与SOX10反式激活MITF启动子,协同上调MITF的表达。作为MITF的一种效应基因,是Kit和E-钙粘蛋白基因的转录抑制因子,影响黑色素细胞的发育。Piebaldism:斑驳病;Tietz:Tietz综合征(Tietz syndrome);WS:Waardenburg综合征(Waardenburg syndrome)。
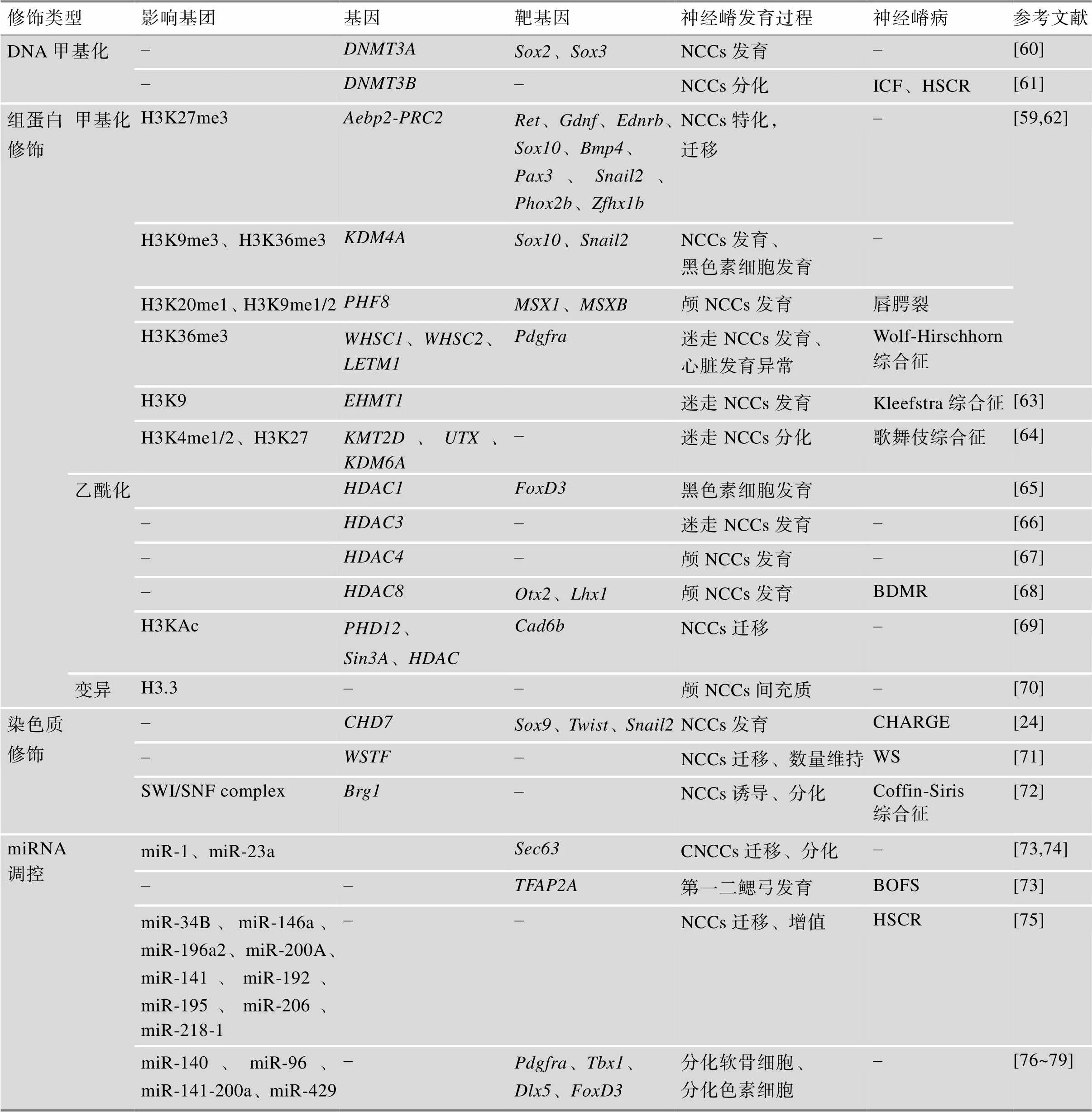
表4 表观遗传修饰对神经嵴发育的调控
BDMR:非综合征性口唇裂及短趾性智力发育迟滞综合征(non-syndromic oral clefts and brachydac-tyly mental retardation syndrome);BOFS:鳃–眼–面部综合征(branchio-oculo-facial syndrome);HSCR:先天性巨结肠症(Hirschsprung disease)。
3.3.1 乙醇:胎儿酒精谱系障碍(fetal alcohol spectrum disorder,FASD)
怀孕期间饮酒现在被认为是一种潜在的致畸风险,可能导致以眼睑裂隙短上唇延长和发育不全为特征的胎儿酒精谱系障碍[82]。怀孕期间过度接触酒精会导致胎儿颅面畸形,占所有酒精导致的出生缺陷的1/3。研究发现将早期胚胎暴露在乙醇中,发现细胞内的酶活性降低,核糖体凋亡,并破坏了神经的发育,其中包括神经元存活、增殖、细胞迁移和突触的形成都受到了影响[83]。取鸡胚胎研究的额外证据表明,酒精能抑制Wnt和Shh信号,从而改变NCCs的迁移和存活[84]。最终导致颅和心脏NCCs数量减少,迁移不流足,引发颅面部畸形甚至先天性心脏病[81]。
3.3.2 维甲酸(retinoic acid,RA)
维生素A胚胎病是一种由子宫内过度暴露于维甲酸引起的NCP,以小耳朵、颌骨发育不全、腭裂和心脏异常为特征,RA已被证明对NCCs的形成至关重要[85,86]。RA摄入量的细微差异都将对NCCs的发展产生重大影响,孕期服用RA药物会损害正常的颅面形态发生。过量的RA也会影响涉及颅骨发育的的表观遗传效应。RA还被发现会导致小鼠的口腔–面部骨折,与两个同源框基因2和表达异常相关[87]。此外,RA信号的改变可导致一种严重的致盲疾病:先天性青光眼[88]。可以肯定的是RA致畸的分子基础是在胚胎期RA刺激p53过度激活,增加了NCCs的凋亡,导致颅骨、心脏甚至骶骨NCCs缺乏[89,90]。
3.3.3 烟雾:香烟、尼古丁、烟草、烟尘
抽烟(尼古丁)也是导致出生类缺陷的重要风险因素,孕妇孕期接触香烟会导致胎儿烟草综合征(fetal tobacco syndrome,FTS),除了呼吸异常和心脏缺陷外,FTS的主要特征是唇腭裂[91]。在非洲爪蟾胚胎和哺乳动物NCCs中进行的一项研究表明,尼古丁和雾化电子烟液的暴露会导致多种出生缺陷,包括正中面部裂隙和中面部发育不全[92]。胚胎暴露在香烟烟雾提取物(一种含有低水平的芳香烃受体(aromatic hydrocarbon receptor,AHR)配体化合物)中,主要通过下调Wnt信号通路的共激活因子R-spondin1影响Wnt通路,该通路是迁移性NCCs的关键信号[93]。皮肤中的所有细胞都表达AHR,如在黑素细胞、朗格汉斯细胞(langerhans cells,LCs)、表皮外层和成纤维细胞等[94]。因此长期暴露在尼古丁下的胎儿,心脏、皮肤等发育都将受到影响。
3.3.4 叶酸(folic acid, FA):甲氨蝶呤(methotrexate, MTX)
叶酸对神经嵴细胞的发育至关重要。在怀孕期,合理的叶酸补充可以防止神经管缺陷、颅面畸形和心脏缺陷的发生[95]。此外,体外和体内实验表明,叶酸水平的异常变化可导致鸡心NCCs的异常迁移和分化。在早期的NCCs特化过程中,正常的口腔发育需要DHFR信号[96]。MTX是FA的拮抗剂,它竞争性地抑制二氢叶酸还原酶(dihydrofolate reductase,DHFR),导致叶酸的缺乏[96,97]。在鸡胚实验中,MTX处理会促使两种主要的叶酸转运蛋白FolR1和RFC1的下调,影响和的正常表达,扰乱了口腔面部组织的形成,推测叶酸可能参与了胎儿口面组织的发育[98]。
4 结语与展望
神经嵴细胞对脊椎动物头部的“精细雕琢”赋予其进化上的绝对优势。在人体内,其形成了复杂有序的神经系统与感观系统,使人类拥有强大的认知和创造能力。然而,当今人们对神经嵴细胞的认知依旧较浅,虽然现有研究对NCCs的形成、迁移和分化进行了解析,并在组织和单细胞维度初步揭示了NCCs多能性机制、细胞命运决定模式和发育进程,但NCCs的发育和命运如何被精准调控、NCCs的迁移路线如何决定、NCCs和周围细胞的如何交互共分化、在时空层面NCCs如何被精准调控以及基因突变影响了NCCs的哪个发育阶段等科学问题仍有待深入揭示。
由于神经嵴细胞发育过程复杂,受到多种信号因子的调节,导致被扰乱的神经嵴细胞与最终发生畸变的组织类型之间匹配困难:即具有相同表型的NCPs可能是由不同NCCs类型的不同发育过程被扰乱所引起,如先天性巨结肠既可由NCCs迁移异常也可由分化异常所导致[75];不同表型的NCPs可能是由同一基因在NCCs不同发育阶段的突变导致的,如SOX10的突变可以在NCCs迁移或分化中分别导致先天性巨结肠或WS[99]。因此神经嵴病的症状具有较大的表型异质性和病因同源性,这给研究NCCs发育过程和NCPs的致病机制带来了严峻考验。精准明晰NCPs的致病突变将有助于实施针对性的治疗和预防手段[100],在基因、蛋白和功能层面的在体外动物实验有助于阐明NCPs的致病机制,并揭示关键治疗靶点[101]。
随着单细胞RNA测序的进展,特别是单细胞多组学方法的快速发展,神经嵴谱系中各类细胞的形成节点和驱动其多能性的机制也逐渐被解析。NCCs单细胞数据证实了之前对神经嵴基因调控网络的认知,并且揭示了许多新的转录调控因子,并发现了新的调控网络(迷走–神经嵴调控网络)[102,103]。同时经典的神经胚层假说也可以验证:即NCCs起源于神经外胚层,具备分化为多种类型细胞的多能性。最新研究揭示NCCs可被Oct4重编程恢复至囊胚期的多能性,进而分化为其他胚层细胞[104],从而推测NCCs不仅仅是“第四胚层”,其多能性甚至要高于内、中和外胚层。Soldatov等[6]利用NCCs的单细胞数据发现NCCs的命运似乎从迁移开始前后就已经被决定,NCCs的命运决定有三个主要阶段:(1)迁移前后的协同激活,(2)迁移过程中的命运逐步偏倚和(3)迁移结束的分化阶段不同的细胞决策。然而,单细胞方法不可避免地失去了空间/解剖位置的信息,空间组学的最新进展将在不久的将来揭示神经嵴细胞命运如何在正确的时间和地点被激活以形成相应的衍生物,为神经嵴细胞的命运和功能带来新的认知,为神经嵴病的病因学揭示和疾病防控及治疗提供关键技术。
附加材料见文章电子版wwww.chinagene.cn。
[1] Bae CJ, Saint-Jeannet JP. Induction and specification of neural crest cells: extracellular signals and transcriptional switches., 2014, 27–49.
[2] Martik ML, Gandhi S, Uy BR, Gillis JA, Green SA, Simoes-Costa M, Bronner ME. Evolution of the new head by gradual acquisition of neural crest regulatory circuits., 2019, 574(7780): 675–678.
[3] Martik ML, Bronner ME. Riding the crest to get a head: neural crest evolution in vertebrates., 2021, 22(10): 616–626.
[4] Hoppler S, Wheeler GN. DEVELOPMENTAL BIOLOGY. It's about time for neural crest., 2015, 348(6241): 1316–1317.
[5] Yuan Y, Loh YHE, Han X, Feng JF, Ho TV, He JZ, Jing JJ, Groff K, Wu AL, Chai Y. Spatiotemporal cellular movement and fate decisions during first pharyngeal arch morphogenesis., 2020, 6(51): eabb0119.
[6] Soldatov R, Kaucka M, Kastriti ME, Petersen J, Chontorotzea T, Englmaier L, Akkuratova N, Yang YS, Häring M, Dyachuk V, Bock C, Farlik M, Piacentino ML, Boismoreau F, Hilscher MM, Yokota C, Qian XY, Nilsson M, Bronner ME, Croci L, Hsiao WY, Guertin DA, Brunet JF, Consalez GG, Ernfors P, Fried K, Kharchenko PV, Adameyko I. Spatiotemporal structure of cell fate decisions in murine neural crest., 2019, 364(6444): eaas9536.
[7] Zhu YL, Crowley SC, Latimer AJ, Lewis GM, Nash R, Kucenas S. Migratory neural crest cells phagocytose dead cells in the developing nervous system., 2019, 179(1): 74–89. e10.
[8] Le Douarin NM, Dupin E. The neural crest, a fourth germ layer of the vertebrate embryo: significance in chordate evolution., 2014, 3–26.
[9] Bolande RP. The neurocristopathies: A unifying concept of disease arising in neural crest maldevelopment., 1974, 5(4): 409–429.
[10] Trainor P, Krumlauf R, Bronner-Fraser M. 19 - Neural Crest Cells. In:Edited by Lanza R, Gearhart J, Hogan B, Melton D, Pedersen R, Thomson J, West M. Burlington: Academic Press, 2004, 219–232.
[11] Vega-Lopez GA, Cerrizuela S, Tribulo C, Aybar MJ. Neurocristopathies: New insights 150 years after the neural crest discovery., 2018, 444 (Suppl 1): S110–S143.
[12] Alkobtawi M, Pla P, Monsoro-Burq AH. BMP signaling is enhanced intracellularly by FHL3 controlling WNT- dependent spatiotemporal emergence of the neural crest., 2021, 35(12): 109289.
[13] Copeland J, Simoes-Costa M. Post-transcriptional tuning of FGF signaling mediates neural crest induction., 2020, 117(52): 33305–33316.
[14] Minoux M, Rijli FM. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development., 2010, 137(16): 2605–2621.
[15] Dyson Y, Holmes A, Li A, Kulesa PM. A chemotactic model of trunk neural crest cell migration., 2018, 56(9): e23239.
[16] Burns AJ, Le Douarin NM. Enteric nervous system development: analysis of the selective developmental potentialities of vagal and sacral neural crest cells using quail-chick chimeras., 2010, 262(1): 16–28.
[17] Szabó A, Mayor R. Mechanisms of neural crest migration., 2018, 52: 43–63.
[18] Simões-Costa M, Bronner ME. Insights into neural crest development and evolution from genomic analysis., 2013, 23(7): 1069–1080.
[19] Reed RJ. Cutaneous manifestations of neural crest disorders (neurocristopathies)., 1977, 16(10): 807–826.
[20] A Vega-Lopez G, J Aybar M. Neurocristopathies: how new discoveries in neural crest research changed our understanding., 2018, 7(2).
[21] Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, Helms JA. Cranial neural crest cells on the move: their roles in craniofacial development., 2011, 155A(2): 270–279.
[22] Xu XP, Wang BQ, Jiang ZY, Chen Q, Mao K, Shi XF, Yan C, Hu JT, Zha Y, Ma C, Zhang J, Guo R, Wang LG, Zhao SQ, Liu HS, Zhang QG, Zhang YB. Novel risk factors for craniofacial microsomia and assessment of their utility in clinic diagnosis., 2021, 30(11): 1045– 1056.
[23] Zhang DC, Ighaniyan S, Stathopoulos L, Rollo B, Landman K, Hutson J, Newgreen D. The neural crest: a versatile organ system., 2014, 102(3): 275–298.
[24] Schulz Y, Wehner P, Opitz L, Salinas-Riester G, Bongers EMHF, van Ravenswaaij-Arts CMA, Wincent J, Schoumans J, Kohlhase J, Borchers A, Pauli S. CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance., 2014, 133(8): 997–1009.
[25] Mort RL, Jackson IJ, Patton EE. The melanocyte lineage in development and disease., 2015, 142(7): 1387.
[26] Kelberman D, Rizzoti K, Lovell-Badge R, Robinson ICAF, Dattani MT. Genetic regulation of pituitary gland development in human and mouse., 2009, 30(7): 790–829.
[27] Tortora C, Meazzini MC, Garattini G, Brusati R. Prevalence of abnormalities in dental structure, position, and eruption pattern in a population of unilateral and bilateral cleft lip and palate patients., 2008, 45(2): 154–162.
[28] Li X, Hu JT, Zhang J, Jin Q, Wang DM, Yu J, Zhang QG, Zhang YB. Genome-wide linkage study suggests a susceptibility locus for isolated bilateral microtia on 4p15. 32-4p16. 2., 2014, 9(7): e101152.
[29] Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata., 2000, 45(3): 175–194.
[30] SooHoo JR, Davies BW, Allard FD, Durairaj VD. Congenital ptosis., 2014, 59(5): 483– 492.
[31] Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du CY, Dixon J, Dixon MJ, Trainor PA. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function., 2008, 14(2): 125–133.
[32] Sakai D, Trainor PA. Face off against ROS: Tcof1/Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development., 2016, 58(7): 577–585.
[33] Selleck MA, Bronner-Fraser M. Origins of the avian neural crest: the role of neural plate-epidermal interactions., 1995, 121(2): 525–538.
[34] Zhang YB, Hu JT, Zhang J, Zhou X, Li X, Gu CH, Liu T, Xie YC, Liu JQ, Gu ML, Wang PP, Wu TT, Qian J, Wang Y, Dong XQ, Yu J, Zhang QG. Genome-wide association study identifies multiple susceptibility loci for craniofacial microsomia., 2016, 7: 10605.
[35] Wilson DN, Doudna Cate JH. The structure and function of the eukaryotic ribosome., 2012, 4(5): a011536.
[36] Narla A, Hurst SN, Ebert BL. Ribosome defects in disorders of erythropoiesis., 2011, 93(2): 144-149.
[37] Selvi R, Mukunda PA. Role of SOX9 in the etiology of Pierre-Robin syndrome., 2013, 16(5): 700–704.
[38] Boudjadi S, Chatterjee B, Sun WY, Vemu P, Barr FG. The expression and function of PAX3 in development and disease., 2018, 666: 145–157.
[39] Li H, Sheridan R, Williams T. Analysis of TFAP2A mutations in Branchio-Oculo-Facial Syndrome indicates functional complexity within the AP-2α DNA-binding domain., 2013, 22(16): 3195–3206.
[40] Dixon J, Trainor PA, Dixon MJ. TCOF1 (treacle) and the treacher-collins syndrome. In: The Molecular Basis of Clinical Disorders of Morphogenesis. Oxford University Press. 2016.
[41] Dauwerse JG, Dixon J, Seland S, Ruivenkamp CAL, van Haeringen A, Hoefsloot LH, Peters DJM, Boers ACD, Daumer-Haas C, Maiwald R, Zweier C, Kerr B, Cobo AM, Toral JF, Hoogeboom AJM, Lohmann DR, Hehr U, Dixon MJ, Breuning MH, Wieczorek D. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome., 2011, 43(1): 20–22.
[42] Engidaye G, Melku M, Enawgaw B. Diamond blackfan anemia: genetics, pathogenesis, diagnosis and treatment., 2019, 30(1): 67–81.
[43] Chang CF, Schock EN, Billmire DA, Brugmann SA. Craniofacial syndromes: etiology, impact and treatment. In: Principles of Developmental Genetics (Second Edition), 2015, 653–676.
[44] Cassina M, Cerqua C, Rossi S, Salviati L, Martini A, Clementi M, Trevisson E. A synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome., 2017, 25(3): 371–375.
[45] Shi JL, Severson C, Yang JX, Wedlich D, Klymkowsky MW. Snail2 controls mesodermal BMP/Wnt induction of neural crest., 2011, 138(15): 3135–3145.
[46] Sánchez-Mejías A, Fernández RM, López-Alonso M, Añtinolo G, Borrego S. New roles of EDNRB and EDN3 in the pathogenesis of Hirschsprung disease., 2010, 12(1): 39–43.
[47] Fontanella B, Russolillo G, Meroni G. MID1 mutations in patients with X-linked Opitz G/BBB syndrome., 2008, 29(5): 584–594.
[48] Ghosh S, Li L, Tourtellotte WG. Retrograde nerve growth factor signaling abnormalities and the pathogenesis of familial dysautonomia., 2021, 16(9): 1795–1796.
[49] Bachetti T, Ceccherini I. Causative and common PHOX2B variants define a broad phenotypic spectrum., 2020, 97(1): 103–113.
[50] Lakhdar Y, El Houda HA, Mounji H, Elfakiri M, Rochdi Y, Moutaouakil A, Raji A. The Tietz syndrome associated with cardiac malformation: a case report with literature review.2021, 37(1): 112.
[51] Pingault V, Zerad L, Bertani-Torres W, Bondurand N. SOX10: 20 years of phenotypic plurality and current understanding of its developmental function., 2021, DOI: 10. 1136/jmedgenet-2021-108105.
[52] Agarwal S, Ojha A. Piebaldism: A brief report and review of the literature., 2012, 3(2): 144–147.
[53] Papakonstantinou E, Bacopoulou F, Brouzas D, Megalooikonomou V, D'Elia D, Bongcam-Rudloff E, Vlachakis D. NOTCH3 and CADASIL syndrome: a genetic and structural overview., 2019, 24: e921.
[54] Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, Sykiotis GP, Gregory LC, Diaczok D, Tziaferi V, Elting MW, Padidela R, Plummer L, Martin C, Feng B, Zhang CK, Zhou QY, Chen HB, Mohammadi M, Quinton R, Sidis Y, Radovick S, Dattani MT, Pitteloud N. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia., 2012, 97(4): E694–E699.
[55] Chacon-Camacho OF, Fuerte-Flores BI, Zenteno JC. TP63 mutation in a patient with acro-dermo-ungual- lacrimal- tooth syndrome: additional evidence of molecular overlap of the ADULT and EEC syndromes., 2016, 170(6): 1635–1638.
[56] Baggiolini A, Varum S, Mateos JM, Bettosini D, John N, Bonalli M, Ziegler U, Dimou L, Clevers H, Furrer R, Sommer L. Premigratory and migratory neural crest cells are multipotent in vivo., 2015, 16(3): 314–322.
[57] Theveneau E, Mayor R. Neural crest cell migration: guidance, pathways, and cell–cell interactions., 2014, 73–88.
[58] Grill C, Bergsteinsdóttir K, Ogmundsdóttir MH, Pogenberg V, Schepsky A, Wilmanns M, Pingault V, Steingrímsson E. MITF mutations associated with pigment deficiency syndromes and melanoma have different effects on protein function., 2013, 22(21): 4357–4367.
[59] Strobl-Mazzulla PH, Bronner ME. Epigenetic regulation of neural crest cells., 2014, 89–100.
[60] Hu N, Strobl-Mazzulla P, Sauka-Spengler T, Bronner ME. DNA methyltransferase3A as a molecular switch mediating the neural tube-to-neural crest fate transition., 2012, 26(21): 2380–2385.
[61] Jin BL, Tao Q, Peng JR, Soo HM, Wu W, Ying JM, Fields CR, Delmas AL, Liu XF, Qiu JX, Robertson KD. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function., 2008, 17(5): 690–709.
[62] Yu C, Yao XM, Zhao JL, Wang P, Zhang Q, Zhao CJ, Yao SH, Wei YQ. Wolf-Hirschhorn syndrome candidate 1 (whsc1) functions as a tumor suppressor by governing cell differentiation., 2017, 19(8): 606–616.
[63] Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Geneviève D, Cormier-Daire V, van Esch H, Fryns JP, Hamel BCJ, Sistermans EA, de Vries BBA, van Bokhoven H. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome., 2006, 79(2): 370–377.
[64] Shpargel KB, Starmer J, Wang CC, Ge K, Magnuson T. UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome., 2017, 114(43): E9046– E9055.
[65] Ignatius MS, Moose HE, El-Hodiri HM, Henion PD. colgate/hdac1 repression of foxd3 expression is required to permit mitfa-dependent melanogenesis., 2008, 313(2): 568–583.
[66] Singh N, Trivedi CM, Lu MM, Mullican SE, Lazar MA, Epstein JA. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract., 2011, 109(11): 1240–1249.
[67] DeLaurier A, Nakamura Y, Braasch I, Khanna V, Kato H, Wakitani S, Postlethwait JH, Kimmel CB. Histone deacetylase-4 is required during early cranial neural crest development for generation of the zebrafish palatal skeleton., 2012, 12(1): 16.
[68] Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8., 2009, 23(14): 1625–1630.
[69] Strobl-Mazzulla PH, Bronner ME. A PHD12-Snail2 repressive complex epigenetically mediates neural crest epithelial-to-mesenchymal transition., 2012, 198(6): 999–1010.
[70] Cox SG, Kim H, Garnett AT, Medeiros DM, An W, Crump JG. An essential role of variant histone H3. 3 for ectomesenchyme potential of the cranial neural crest., 2012, 8(9): e1002938.
[71] Yoshimura K, Kitagawa H, Fujiki R, Tanabe M, Takezawa S, Takada I, Yamaoka I, Yonezawa M, Kondo T, Furutani Y, Yagi H, Yoshinaga S, Masuda T, Fukuda T, Yamamoto Y, Ebihara K, Li DY, Matsuoka R, Takeuchi JK, Matsumoto T, Kato S. Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF)., 2009, 106(23): 9280–9285.
[72] Kosho T, Okamoto N, Coffin-Siris Syndrome International Collaborators. Genotype-phenotype correlation of Coffin- Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A., 2014, 166C(3): 262–275.
[73] Wang DY, Weng YJ, Guo SY, Qin WH, Ni JL, Yu L, Zhang YX, Zhao QS, Ben JJ, Ma JQ. MicroRNA-1 regulates NCC migration and differentiation by targeting sec63., 2019, 15(12): 2538–2547.
[74] Wei AY, Zhao PH, Xia JL, Wang Q, Du XH. MicroRNA- 23a is required for the migration and differentiation of cranial neural crest cells of zebrafish., 2017, 5(1): 7–13.
魏安瑶, 赵鹏辉, 夏景兰, 王强, 杜兴华. microRNA- 23a调控斑马鱼颅神经嵴细胞迁移和分化. 发育医学电子杂志, 2017, 5(1): 7–13.
[75] Torroglosa A, Alves MM, Fernández RM, Añtinolo G, Hofstra RM, Borrego S. Epigenetics in ENS development and Hirschsprung disease., 2016, 417(2): 209–216.
[76] Eberhart JK, He XJ, Swartz ME, Yan YL, Song H, Boling TC, Kunerth AK, Walker MB, Kimmel CB, Postlethwait JH. MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis., 2008, 40(3): 290–298.
[77] Gao S, Moreno M, Eliason S, Cao HJ, Li X, Yu WJ, Bidlack FB, Margolis HC, Baldini A, Amendt BA. TBX1 protein interactions and microRNA-96-5p regulation controls cell proliferation during craniofacial and dental development: implications for 22q11. 2 deletion syndrome., 2015, 24(8): 2330–2348.
[78] Itoh T, Nozawa Y, Akao Y. MicroRNA-141 and -200a are involved in bone morphogenetic protein-2-induced mouse pre-osteoblast differentiation by targeting distal-less homeobox 5., 2009, 284(29): 19272–19279.
[79] Yan B, Liu B, Zhu CD, Li KL, Yue LJ, Zhao JL, Gong XL, Wang CH. MicroRNA regulation of skin pigmentation in fish., 2013, 126(Pt 15): 3401–3408.
[80] Sato TS, Handa A, Priya S, Watal P, Becker RM, Sato Y. Neurocristopathies: Enigmatic Appearances of Neural Crest Cell-derived Abnormalities., 2019, 39(7): 2085–2102.
[81] Cerrizuela S, Vega-Lopez GA, Aybar MJ. The role of teratogens in neural crest development., 2020, 112(8): 584–632.
[82] Fainsod A, Kot-Leibovich H. Xenopus embryos to study fetal alcohol syndrome, a model for environmental teratogenesis., 2018, 96(2): 77–87.
[83] Tolosa EJ, Fernández-Zapico ME, Battiato NL, Rovasio RA. Sonic hedgehog is a chemotactic neural crest cell guide that is perturbed by ethanol exposure., 2016, 95(3/5): 136–152.
[84] Flentke GR, Baulch JW, Berres ME, Garic A, Smith SM. Alcohol-mediated calcium signals dysregulate pro- survival Snai2/PUMA/Bcl2 networks to promote p53-mediated apoptosis in avian neural crest progenitors., 2019, 111(12): 686–699.
[85] Mulder GB, Manley N, Grant J, Schmidt K, Zeng W, Eckhoff C, Maggio-Price L. Effects of excess vitamin A on development of cranial neural crest-derived structures: a neonatal and embryologic study., 2000, 62(4): 214–226.
[86] Mondal D, Shenoy SR, Mishra S. Retinoic acid embryopathy., 2017, 7(4): 264–265.
[87] Zhao Y, Guo YJ, Tomac AC, Taylor NR, Grinberg A, Lee EJ, Huang S, Westphal H. Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene lhx8., 1999, 96(26): 15002–15006.
[88] Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view., 2008, 9(7): 541–553.
[89] Melnik BC. Overexpression of p53 explains isotretinoin's teratogenicity., 2018, 27(1): 91–93.
[90] Melnik BC. Apoptosis may explain the pharmacological mode of action and adverse effects of isotretinoin, including teratogenicity., 2017, 97(2): 173–181.
[91] Bronner ME, LeDouarin NM. Development and evolution of the neural crest: an overview., 2012, 366(1): 2–9.
[92] Kennedy AE, Kandalam S, Olivares-Navarrete R, Dickinson AJG. E-cigarette aerosol exposure can cause craniofacial defects in Xenopus laevis embryos and mammalian neural crest cells., 2017, 12(9): e0185729.
[93] Sanbe A, Mizutani R, Miyauchi N, Yamauchi J, Nagase T, Yamamura KI, Tanoue A. Inhibitory effects of cigarette smoke extract on neural crest migration occur through suppression of R-spondin1 expression via aryl hydrocarbon receptor., 2009, 380(6): 569–576.
[94] Esser C, Bargen I, Weighardt H, Haarmann-Stemmann T, Krutmann J. Functions of the aryl hydrocarbon receptor in the skin., 2013, 35(6): 677–691.
[95] Li JJ, Shi Y, Sun J, Zhang YF, Mao BY. Xenopus reduced folate carrier regulates neural crest development epigenetically., 2011, 6(11): e27198.
[96] Silveira AB, Laranjeira ABA, Rodrigues GOL, Leal PC, Cardoso BA, Barata JT, Yunes RA, Zanchin NIT, Brandalise SR, Yunes JA. PI3K inhibition synergizes with glucocorticoids but antagonizes with methotrexate in T-cell acute lymphoblastic leukemia., 2015, 6(15): 13105–13118.
[97] Rajagopalan PTR, Zhang ZQ, McCourt L, Dwyer M, Benkovic SJ, Hammes GG. Interaction of dihydrofolate reductase with methotrexate: ensemble and single- molecule kinetics., 2002, 99(21): 13481–13486.
[98] Alata Jimenez N, Torres Pérez SA, Sánchez-Vásquez E, Fernandino JI, Strobl-Mazzulla PH. Folate deficiency prevents neural crest fate by disturbing the epigenetic Sox2 repression on the dorsal neural tube., 2018, 444 Suppl 1: S193–S201.
[99] Zhang H, Chen HS, Luo HJ, An J, Sun L, Mei LY, He CF, Jiang L, Jiang W, Xia K, Li JD, Feng Y. Functional analysis of Waardenburg syndrome- associated PAX3 and SOX10 mutations: report of a dominant-negative SOX10 mutation in Waardenburg syndrome type II., 2012, 131(3): 491–503.
[100] Chen Q, Zhao Y, Shen G, Dai J. Etiology and pathogenesis of hemifacial microsomia., 2018, 97(12): 1297–1305.
[101] Gouignard N, Andrieu C, Theveneau E. Neural crest delamination and migration: looking forward to the next 150 years., 2018, 56(6-7): e23107.
[102] Zalc A, Sinha R, Gulati GS, Wesche DJ, Daszczuk P, Swigut T, Weissman IL, Wysocka J. Reactivation of the pluripotency program precedes formation of the cranial neural crest., 2021, 371(6529): eabb4776.
[103] Morarach K, Mikhailova A, Knoflach V, Memic F, Kumar R, Li W, Ernfors P, Marklund U. Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing., 2021, 24(1): 34–46.
[104] Artinger KB, Monsoro-Burq AH. Neural crest multipotency and specification: power and limits of single cell transcriptomic approaches., 2021, 10: 38.
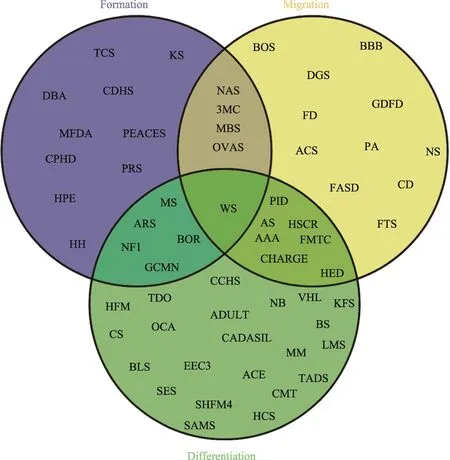
附图1 NCPs分类统计Venn图
Supplementary Fig. 1 Venn chart of NCPs classification
根据受累神经嵴细胞发育过程对神经嵴病进行分类,重叠部分清楚地显示了某些NCPs归因于多个过程发育受阻。
Abbreviations of NCPs described in this article: TCS: Treacher Collins syndrome Treacher Collins综合征;KS: Kallmann Syndrome Kallmann综合征;PRS: Pierre Robin Sequence皮埃尔·罗宾序列;HPE: holoprosencephaly前脑无裂畸形;HH: hypogonadotropic hypogonadism 性腺功能减退症;MBS: Moebius Syndrome莫比乌斯综合征;3MC: 3MC Syndrome 3MC综合征;ADULT: ADULT Syndrome ADULT综合征;AEC: AEC Syndrome AEC综合征;AS: Alagille Syndrome Alagille综合征;BOR: Branchio-Oto-Renal Syndrome鳃-耳-肾综合征;CADASIL: Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy大脑常染色体显性动脉病变伴皮层下梗死和白质脑病;CDHS: Craniofacial-Deafness-Hand Syndrome 颅面-耳聋-手综合征;GCMN: Giant Congenital Melanocytic Nevi巨大先天性黑素细胞痣;NF1: Neurofibromatosis I Ⅰ型神经纤维瘤;WS: Waardenburg syndrome瓦登伯格综合征;CED: Cranioectodermal dysplasia/Sensenbrenner syndrome颅骨外胚层发育不良/Sensenbrenner综合征;EEC3: EEC3 syndrome EEC3综合征;EVC: Ellis-van Creveld syndrome软骨外胚层发育不良;FASD: Fetal Alcohol Spectrum Disorder胎儿酒精谱系障碍;GDFD: Growth Retardation, Developmental Delay, and Facial Dysmorphism生长迟缓,发育迟缓,面部畸形;HCS: Hajdu-Cheney Syndrome Hajdu-Cheney综合征;FS: Klippel-Feil Syndrome先天性短颈综合征;LMS: Limb-Mammary Syndrome四肢-乳腺综合征;MFDA: Mandibulofacial Dysostosis with Alopecia颌面部发育不良伴脱发;MS: Multiple sclerosis多发性硬化症;NAD: Nager Acrofacial Dysostosis Nager综合征;NS: Noonan syndrome Noonan综合征;PA: Peters Anomaly Peter异常/前段发育不良;PHACE: PHACE Syndrome PHACE综合征;SAMS: SAMS disorder SAMS异常;SHFM4: Split-hand/split-foot malformation type 4裂手/裂脚畸形4型;TDO: Tricho-dento-osseous syndrome毛齿骨综合征;22q11: 22q11.2 Deletion Syndrome (DGS) 22q11.2缺失综合症;AAA: Achalasia- Addisonianism-Alacrima Syndrome AAA综合征;ACS: Auriculo Condylar Syndrome耳廓-髁状突综合征;ARS: Axenfeld- Rieger Syndrome Axenfeld-Rieger综合征;BLS: Bamforth-Lazarus syndrome Bamforth-Lazarus综合症;BOFS: Branchio-Oculo-Facial Syndrome 鳃-眼-面部综合征;BS: Binder Syndrome Binder综合征;CCHS: Congenital Central Hypoventilation Syndrome先天性中枢性通气不足综合征;CHARGE: CHARGE syndrome CHARGE综合征;CMT: Charcot-Marie-Tooth and Deafness Syndrome腓骨肌萎缩症和耳聋综合症;CS: Craniosynostosis颅缝早闭;DBA: Diamond Blackfan Anemia先天性再生障碍性贫血;FD: Familial dysautonomia家族性自主神经障碍;FMTC: Familial medullary thyroid carcinomas家族性甲状腺髓样癌;HFM: Hemifacial microsomia半侧颜面短小.
Research progress on neural crest cells and neurocristopathies and its pathogenesis
Zhuoyuan Jiang1, Yan Zha1, Xiaofeng Shi3,4, Yongbiao Zhang2,3,4
Neural crest cells (NCCs) are multipotent progenitor cells unique to vertebrates, and they have the ability to differentiate into a variety of cells, such as chondrocytes, neurons, and melanocytes. The formation, migration, and differentiation of NCCs are tightly regulated, and the disruption of NCC development results in abnormal embryo development. Neurocristopathies (NCPs) refer to a group of diseases that develop in response to abnormal development of NCCs. NCPs are of various types and exhibit complex phenotypes, which can affect many parts of the human body, such as the craniofacial structure, heart, intestine, and skin. NCPs negatively impact the physical function and mental health of the affected patients. NCPs account for one third of the defects in children with birth defects. Genetic factors are the main risk factors for NCPs, but environmental factors and abnormal gene-environment interactions can also lead to the development of NCPs. In this review, we introduce NCCs, NCPs, and their pathogenesis, so as to provide a reference point for a systematic understanding of NCPs and NCC development, and to provide scientific support for understanding the etiology of NCPs and their effective prevention and control.
neural crest cells (NCCs); neurocristopathy; genetic regulation network; risk factors; pathogenesis
2021-09-29;
2021-12-11;
2022-01-04
国家自然科学基金(编号:82171844,81970898)和北京市自然科学基金(编号:7204273)资助[Supported by the National Natural Science Foundation of China (Nos. 82171844, 81970898) and Beijing Natural Science Foundation Project (No. 7204273)]
蒋卓远,在读硕士研究生,专业方向:基础医学。E-mail: SY1910307@buaa.edu.cn
张永彪,博士,副研究员,研究方向:生物信息学。E-mail: zhangyongbiao@buaa.edu.cn
10.16288/j.yczz.21-253
(责任编委: 何淑君)
