双金属催化剂用于烯烃氢甲酰化反应的研究进展
2022-03-08支晓彤姜伟丽李继聪周广林周红军
支晓彤,姜伟丽,李继聪,王 岩,周广林,周红军
(中国石油大学(北京) 新能源与材料学院,北京 102249)
氢甲酰化反应即烯烃与合成气(CO + H2)在过渡金属催化剂的作用下生成醛或醇类产物的过程,是工业上生产醛类产品最主要的方法,也是迄今为止最重要的均相催化反应之一[1,2]。据统计,全球通过氢甲酰化反应产出的醛已达千万吨级规模[3]。该反应主要采用铑(Rh)或钴(Co)的有机化合物作为催化剂[4]。羰基钴是最早发现的具有高活性的催化剂,但由于其对操作环境的要求较高,随后人们又陆续开发了羰基钴-膦催化剂、油溶性铑膦配合物催化剂以及水溶性铑膦催化剂。由于铑膦催化剂具有高活性和高正异构比,且反应条件更加温和,因此目前工业上大多采用铑膦催化体系[5-10]。
但Rh催化剂的缺点是Rh的价格昂贵。为了降低催化剂成本,一方面研究者积极开发易分离的催化体系,以减少传统均相体系中因蒸馏分离造成的铑催化剂的失活及损失,如水/有机两相催化剂及负载型催化剂,但前者对烯烃的水溶性要求较高,水溶性差的长链烯烃往往催化效果不理想,而且两相传质问题也一直没有得到很好地解决[11];而后者的最大问题是活性中心在载体上的稳定性不足,常导致金属活性中心的脱落,限制了其应用[12,13];另一方面,有研究者一直在尝试开发其他廉价过渡金属配合物催化剂。经研究发现,元素周期表的第八族元素中许多金属的羰基配合物对烯烃的氢甲酰化反应都有催化作用,其催化活性顺序为:Rh > Co > Ir,Ru > Os > Pt > Pd > Fe > Ni[14]。
自Muetterties提出多金属团簇催化可获得高活性催化剂[15-18]以来,开发双金属或多金属配合物以获得与Rh催化性能相当的催化剂引起了研究者的很大兴趣。研究发现,在催化体系中加入第2种金属可能会有利于催化活性的提升,而且在某些情况下,双金属(或多金属)体系的金属中心比具有相同结构的单金属中心更为活跃,这种现象通常归因于两个金属中心之间的协同作用[19]。故此,本文综述了用于氢甲酰化反应的Co系双金属催化剂和Rh系双金属催化剂的研究进展,以期为相关研究提供参考。
1 Co系双金属催化剂
在上世纪四十年代使用的羰基钴催化剂中,发现不论何种形式的钴,如碳酸钴、氧化钴等,在氢甲酰化条件下都会转化为氢羰基钴活性物种[20]。Heck等[21]最早对四羰基钴催化乙烯的反应机理进行了研究,他们提出了反应式(1)~反应式(3)所示的反应路径:

在该反应路径中初次涉及了双金属催化反应机理,即RCH2CH2COCo(CO)4与HCo(CO)4通过分子间氢化物转移导致醛的还原消除和Co2(CO)8的生成。目前工业上对于长链烯烃的氢甲酰化反应依然会采用均相钴膦催化体系,用减压蒸馏对其进行循环浓缩[22],在浓缩过程中由于氢甲酰化产物高碳醛(醇)的沸点高,容易导致Co和膦配体的损失。工业生产中采用定期补充催化剂的方法来弥补催化活性的下降;在实验室研究中,研究者探究了Co催化体系中加入第2种金属来提高Co催化剂的稳定性及活性。其中,Co基双金属催化剂在烯烃氢甲酰化方面的应用主要包括Co-Rh、Co-Ru等。
1.1 Co-Rh双金属催化剂
在氢甲酰化反应领域中,Co与Rh的催化活性是最高的,且Rh的活性高于Co。但Rh价格昂贵, Co价廉易得,因此Co-Rh双金属催化剂引起了研究者的关注。对于Co-Rh双金属催化剂的研究大多集中于多相催化体系中的负载型催化剂,即将金属Co和Rh先后负载到固体材料上,以达到易分离的目的。常用的载体有二氧化硅(SiO2)、分子筛、金属氧化物、水滑石等。
Huang等[23]首先将RhCl3/SiO2在空气中673 K焙烧5 h,将其还原成羰基铑([Rh(CO)2Cl]2和[Rh(CO)2Os]2的混合物),随后在室温下用Co2(CO)8的正己烷溶液浸渍该SiO2负载的混合物,最后在623 K下H2还原后得到高活性Rh-Co/SiO2催化剂。该催化剂在乙烯的氢甲酰化反应中,对含氧化合物的选择性是RhCl3/SiO2催化剂的19倍。作者认为,高催化活性可归因于[Rh(CO)2Os]2在高温下与Co原子相互作用形成高分散的双金属Co-Rh位。易敏等[24]制备了PPh3-Rh-Co/SiO2双金属配合物催化剂,并用于异丁烯氢甲酰化反应考察了催化剂的性能,结果表明,异丁烯与单金属Rh-PPh3/SiO2或Co-PPh3/SiO2催化剂反应5 h后,转化率分别为44.7%和30.2%,目标产物异戊醛的选择性只有77.4%和38.5%;而Co-Rh双金属催化剂上的活性和选择性都显著提高,相同反应条件下异丁烯转化率达到了62.0%,异戊醛的选择性增加到89.3%,且没有副产物特戊醛的生成;同时,其采用程序升温技术研究了该双金属催化剂的协同作用,发现Co和Rh之间的协同作用可以促进二者在载体表面的分散,还会影响活性金属中心周围的电子密度,使得活性中心M-CO键更活泼,从而提高了催化活性。
Ma等[25]研究了Co-Rh双金属催化体系在双环戊二烯(DCPD)氢甲酰化制备二甲酰三环癸烷(DFTD)反应中的应用。为了提高DFTD的选择性,其采用浸渍法制备了MCM-41担载的Rh基催化剂(M-Rh/MCM-41,其中M = Co、Fe、Cu、Ni),经对比发现, Co-Rh双金属催化剂的活性最高,而且无论Co负载量多少,都能在DCPD几乎完全转化的基础上,将产物DFTD的选择性大大提高,其中2%Co-2%Rh/MCM-41(2%为质量分数)催化剂可将DFTD的选择性由8.7%提高到87.3%,在循环使用5次之后,选择性依然可达85.0%。作者认为这可能是由于催化剂表面存在Co改性的高活性Rh物种所致。与单金属Rh催化剂相比,单甲酰三环癸烷(MFTD)选择性则由87.0%降至6.0%,这可能是由于Rh与其他金属电子的共同变化影响了反应物与产物在反应过程中的相互作用。2017年,Ma等[26]又以Na2CO3为沉淀剂,以RhCl3、Co(NO3)2和Fe(NO3)3为原料采用共沉淀法制备了不同Co负载量的Co-Rh/Fe3O4催化剂。结果显示,当Co与Rh的质量比为2:1时,DFTD的选择性提高到90.6%,比MCM-41为载体的双金属催化剂活性更好,而且由于铁的磁性使得催化剂更容易回收利用。关于Co-Rh相互作用的分析与先前的研究结果相似,其DTA表征结果进一步表明了Co导致产物分布变化的原因,即添加Co改变了Rh-P键相互作用强度,而其之前的研究发现,强的Rh-P键对生成DFTD有利,弱的Rh-P键有利于生成MFTD。Ning等[27]研究了SiO2负载的Co-Rh双金属催化剂对乙烯气相氢甲酰化反应的催化性能,结果表明,Co-Rh双金属催化剂比Rh/SiO2催化剂的产率提高了78%,丙醛选择性由38%提高至46%。TEM表征结果表明,Co的加入增加了活性中心的暴露量,从而提高了Co-Rh双金属催化剂的活性。
在多相催化剂的制备过程中,水滑石(HT)是一种常用的催化剂载体。Sharma等[28]采用原位溶胶-凝胶法将Rh3+引入到α-Co(OH)2+xHT型材料中,成功制备了基于水滑石型材料的多相双金属Co-Rh催化剂(粒径约1 mm),并分别以C6、C7、C8以及C10烯烃为底物,研究了催化剂的性能,同时对该催化剂进行了TEM表征,结果如图1所示。
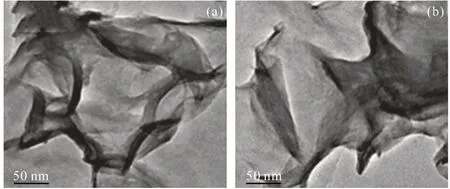
图1 钴铑水滑石基材料负载前(a)和负载后(b)的HR-TEM图[28]Fig.1 HR-TEM images of cobalt-rhodium hydrotalcite based materials (a) before and (b) after loading[28]
由图1可知,载体表面光滑,无Rh粒子,表明Rh在α-Co(OH)2+xHT型材料中以同晶取代形式存在,使得催化剂更稳定;该催化剂的氢甲酰化结果表明,在温度为100 °C、V(CO):V(H2) = 1:1、总压5 MPa的最佳反应条件下持续反应6 h,烯烃的转化率均在96%~99%,C7、C8以及C10醛的选择性达到96%。回收使用4次之后,催化剂上Co和Rh的含量依然保持恒定,转化率仍在90%以上,但醛的选择性由于异构化产物以及二次产物的增加而大大降低。
1.2 Co-Ru和Co-Ni双金属催化剂
负载型Co-Ru催化剂由于可显著提高费托合成和烯烃氢甲酰化反应中的氧化产物生成速率受到研究者的关注[29]。2001年,Huang等[30]研究了SiO2负载的Co-Ru催化体系,并将其与Co-Rh/SiO2催化剂作对比,考察了Co-Ru双金属催化剂的可行性。通过对比金属盐和金属羰基化合物两种催化剂前驱体对氧化产物选择性的影响发现,由Co、Rh羰基化合物衍生得到的催化剂更有利于氧化产物的生成。在Co与Ru原子比为3:1时,氧化产物选择性为47%,且在反应的92 h内,对含氧化合物的催化活性几乎保持不变。由金属氯化物衍生得到的催化剂活性最低,为0.62 mol/(mol⋅min),其认为,这是由于氯化物在含氧化合物形成中的有害作用以及金属氯化物制备的催化剂金属分散性较低所致;在操作条件相同的情况下,Co-Rh 催化剂具有更高的活性和产物选择性(氧化产物选择性为65%),但由于Ru的价格大约为Rh的1/10,而且所研究的含Ru和Co的催化剂稳定性良好,因此认为,Ru基催化剂可能比Rh基催化剂更具实用价值。
邱介山等[31]采用等体积浸渍法制备了碳纳米管(CNTs)负载的Co/CNTs、Co-Ru/CNTs催化剂,并考察了其在1-辛烯氢甲酰化反应中的催化性能。结果显示,与单金属Co催化剂相比,双金属催化剂的转化率由23.30%提高至70.97%,选择性也由48.10%提高至65.75%,这表明Ru、Co金属之间存在某种协同作用。催化剂的TEM表征结果如图2所示。

图2 CNTs及其负载金属催化剂的TEM图[31]Fig. 2 TEM images of CNTs and their supported metal catalysts[31]
从图2可以看出,少量Ru的加入可以增加Co活性位点的密度,使Co在CNTs上分布更均匀,并且可以促进Co的还原,使催化剂在反应过程中不易积炭,提高催化剂稳定性;但过量的Ru会覆盖在Co粒子表面,降低金属Co的催化活性。
此外,赵天生等[32]采用超声辅助浸渍法制备了SBA-15分子筛负载的Co-Ni-B非晶态催化剂,该催化剂在1-辛烯氢甲酰化反应中表现出优异的催化性能,其认为,Co-B中引入Ni一方面促进了Co分散,另一方面增加了电子密度,使得负载Co-Ni-B的氢甲酰化催化性能更佳。
Co催化剂具有价格低廉、对原料中毒物不敏感以及在高碳烯烃的氢甲酰化反应中可使产物醛进一步生成碳数加一的醇等优点,但也存在活性较低的问题。尽管如此,工业上仍主要采用Co基催化剂作为高碳烯烃氢甲酰化反应催化剂[33]。从Co系双金属催化剂的研究结果来看,双金属的协同作用增加了催化体系的活性及稳定性,使其更适用于低碳烯烃的氢甲酰化反应;且负载型双金属催化剂解决了均相反应分离难的问题,在保证活性的同时还降低了膦配体的用量。但双金属催化剂的研究由于还存在负载型催化剂的再生性能差、反应过程中活性中心易脱落以及制备过程复杂等难题未得到有效解决,目前还停留在实验室研究阶段。
2 Rh系双金属催化剂
上世纪七十年代初期,Wilkinson等研究发现了活性比Co催化剂高数百倍的Rh催化剂HRh(CO)2(PPh3)2,这也是目前工业上应用最广泛的氢甲酰化催化剂[34]。与羰基钴催化剂相比,这种催化剂的反应条件更加温和,活性也更高,主要存在的问题是Rh成本太高,另外反应过程中膦配体容易脱落,常需要使用过量的膦配体[4]。为此,在低成本的推动下,研究者在铑配合物催化剂体系中引入第2种金属制备双金属催化剂,期望能够通过双金属的协同作用来达到与单金属铑配合物催化剂相当的活性,该方法因加入的Rh减少而降低了成本,同时还可增加催化剂的稳定性。目前研究中以Rh-Rh、Rh-Co双核催化剂为主,其次是Rh与其他过渡金属的组合。
2.1 Rh-Rh双金属催化剂
Rh-Rh双金属催化剂虽不能降低贵金属Rh的用量,却可以增加Rh的稳定性。研究者对于Rh-Rh双金属配合物的研究大多聚焦于催化剂的合成及性能。1993年Broussard等[35]利用双核四膦配体的内外消旋非对映异构体结构(1r和1m,见图3),制备了催化效果与工业羰基铑膦络合物催化剂相当的均相双金属铑-降冰片二烯配合物,并将其用于催化丙酮溶剂中己烯的氢甲酰化反应。1r或1m与[Rh(nbd)2](BF)4(nbd代表降冰片二烯)反应生成双金属络合物2r([Rh2(nbd)2(Et,Ph-P4)](BF4)2),其对己烯生成醛的转化率达到85%,正异比达到27.5,与商用催化剂相当;内消旋配合物2m由于结构差异,反应时需要更高的能量,导致催化速率变差;2r与CO和H2作用得到活性物种3r,从图3中可以看到其结构中并没有Rh-Rh键,这使得Rh双金属配合物的制备更容易,与单金属铑膦配合物相比,3r中由于螯合物和供电子的Et、Ph-P4膦配体与Rh中心有足够强的配位,不需要过量的膦配体来保持其稳定性和选择性。
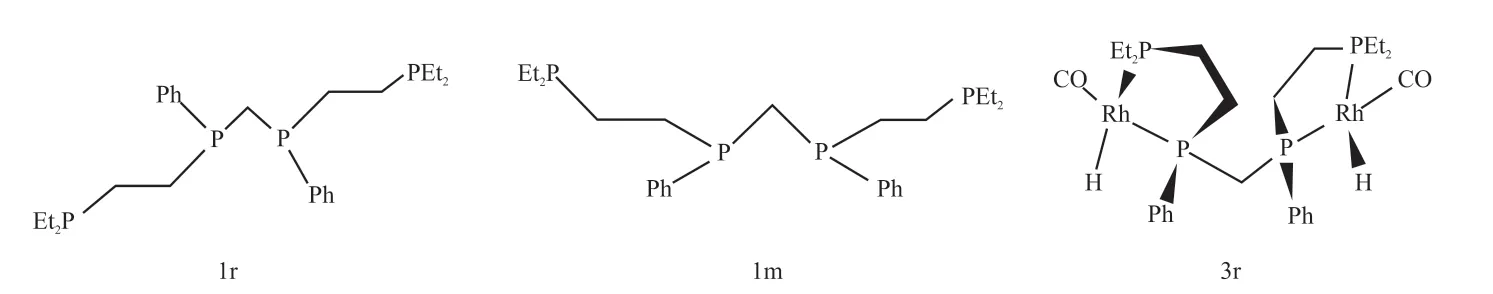
图3 双核四膦配体1r、1m以及双金属铑-降冰片二烯配合物3r的结构[35]Fig. 3 Structures of dinuclear tetraphosphine ligand 1r, 1m and bimetal rhodium-norborndiene complex 3r[35]
Vasam等[36]采用7-氮杂吲哚作为桥联配体,合成了单核Rh(I)-膦催化剂(2)、双核Rh(I)-膦催化剂(1)和(3)~(8),如图4所示,并将其用于1-己烯与十二烯的氢甲酰化反应。结果显示,与单核铑催化剂相比,其他双核[Rh(μ-azi)(CO)(L)]2(azi =7-氮杂吲哚盐,L为膦配体)催化剂都是有效的高碳烯烃氢甲酰化催化剂。在水+甲苯体系中,在反应温度为80 °C、CO 和 H2总压为2.5 MPa的反应条件下,1-己烯转化率为73.7%~96.0%,醛选择性70%~95%,线性醛与支链醛的比例(l/b)为4.8~9.6;十二烯转化率63.9%~92.1%,醛选择性60%~91%,l/b为2.4~5.4;不同条件下催化剂活性顺序均为(4) > (6) > (7) > (1) > (5);作者还发现膦配体的亲水性越强、吸电子基团越多,则催化剂活性越高。具有缺电子和释放环体系的7-氮杂吲哚甲酸型双核间隔基有助于减少Rh(I)与CO的π-反键效应,提高催化剂稳定性,避免了膦配体的过量使用。

图4 以7-氮杂吲哚为配体的单核Rh(I)-膦催化剂(2)和双核Rh(I)-膦催化剂(1)、(3)~(8)的化学式[36]Fig. 4 Chemical formula of mononuclear Rh(I)-phosphine catalyst (2) and binuclear Rh(I)-phosphine catalyst (1),(3)~(8) with 7-azazindole as ligands[36]
2017年Govender等[37]基于水杨醛亚胺支架合成了两个单核铑配合物Rh(I)、Rh(III) 催化剂和一个双金属Rh(I)-Rh(III)混合价配合物催化剂,结构如图5所示,将其用于催化1-辛烯氢甲酰化反应。结果表明,当V(CO):V(H2) = 1:1时,在温度为75~90 °C和压力为3~4 MPa范围内改变温度和压力,3种催化剂对1-辛烯的转化率均可达到90%以上,醛产率也可达80%以上;双金属铑催化剂的TON值(每摩尔催化剂单位活性中心上底物的转化数)为3797~4616 s-1,是两个单核铑催化剂的两倍,表明双金属铑催化剂具有比单核铑催化剂更加优异的活性。通过循环伏安法研究发现,双金属铑催化剂中Rh(III)的存在影响了Rh(I)的氧化电位,可归因于化合物中Rh(I)和Rh(III)金属中心的长程共轭相互作用。

图5 基于水杨醛亚胺支架的3种催化剂催化1-辛烯氢甲酰化反应示意[37]Fig.5 Illustration of hydroformylation of 1-octene catalyzed by three catalysts based on salicylaldeimide scaffolds[37]
2.2 Rh-Fe双金属催化剂
对于Rh-Fe双金属催化剂的研究,主要集中于Rh-二茂铁双金属配合物体系。2018年Matsinha等[38]合成了两种新的二茂铁酰亚氨基吡啶配体(图6中的L1和L2),将其分别与[RhCl(COD)]2和[RuCl2(p-cymene)]2金属前体反应,制备了Rh(I)-Fe(II)和Ru(II)-Fe(II)均相双金属催化剂,并研究了其对于1-辛烯的氢甲酰化反应的催化活性。结果显示,Rh-二茂铁配合物(见图6中的1和4)具有更好的催化活性,在压力为3 MPa、温度为95 °C的条件下,转化率可达98.33%,醛产率达100%,支链醛选择性63%;压力相同时,在温度为75~95 °C范围内升高温度更有利于支链醛的生成,且催化活性也较高,这可能是由于高温有利于催化剂活性物种的形成,使得1-辛烯在高温下异构化增加,从而提高了支链醛的选择性。而Ru-二茂铁配合物(图6中的2和3)活性较差,反应需要在更高的温度和压力下才能得到68.00%的1-辛烯转化率。
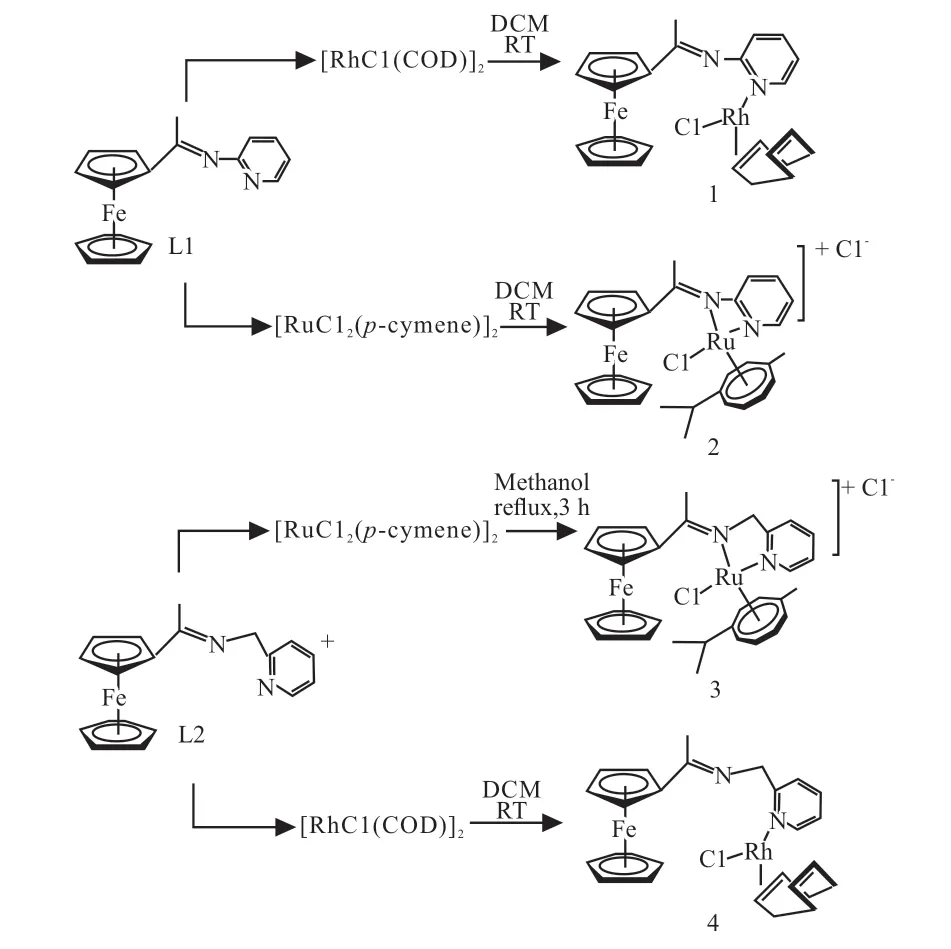
图6 催化剂Rh-二茂铁配合物(1和4)与Ru-二茂铁配合物(2和3)的合成路线[38]Fig. 6 Synthesis routes of rhodium-ferrocene complex (1, 4)and ruthenium-ferrocene complex (2, 3) [38]
2019年Breckwoldt等[39]研究了希夫碱衍生的Rh(I)和Rh(I)-二茂铁催化剂催化7-十四烯的氢甲酰化反应,如图7所示。研究结果显示,两种催化剂均存在诱导期,且在较高温度(> 75 °C)下才表现出更好的催化活性,这可能是由于较低温度下N^O螯合配体与Rh中心的强配位抑制了与金属内部双键的活化;在95 °C、4 MPa的反应条件下,二者都达到了94%~97%的转化率以及88%~92%的醛选择性;同时观察到,与单金属催化剂相比,在温和条件下(85 °C)使用二茂铁基配体的催化剂活性有所提高,这可能是由于二茂铁充当了电子储层,从而通过共轭亚胺π电子体系稳定了催化活性Rh中心周围的电子密度。这种效应可以降低反应的活化能,从而提高反应速率;此外,双金属催化剂在循环使用3次之后仍保持活性。Pogrzeba等[40]研究了以Rh-二茂铁为催化剂的7-十四烯氢甲酰化反应动力学,通过计算得到氢甲酰化反应的活化能为62 kJ/mol,与工业上膦改性的铑催化剂体系的活化能相当。作者认为,该动力学模型可作为异双金属催化剂烯烃氢甲酰化反应器模型开发的起点,通过优化茂金属的吸电子取代基和供电子取代基,这种异双金属催化体系可以用于提高工业环境中Rh的有效性。

图7 希夫碱衍生的Rh(I)催化剂(1)和Rh(I)-二茂铁催化剂(2)催化7-十四烯氢甲酰化反应示意[39]Fig.7 Hydroformylation of 7-tetradecene catalyzed by Rh(I)catalyst (1) and Rh(I)-ferrocene catalyst (2) derived from Schiff base[39]
2.3 Rh与其他金属组合的双金属催化剂
除上述铑Rh系双金属催化剂外,研究者对于Rh与其他金属组合的双金属催化体系也进行了大量 研 究,主 要 有Rh-Mn、Rh-Ni、Rh-Au以 及Rh-Ru等。Li等[41]以未改性的铑和锰羰基为催化剂前驱体,在正己烷溶剂(T≈ 298 K,p(CO) = 1.0~4.0 MPa,p(H2) = 0.5~2.0 MPa)中,研究了3,3-二甲基丁烯低温氢甲酰化制备4,4-二甲基戊醛过程中双金属协同作用的起源,发现在同时使用两种金属的实验中,催化剂催化效率显著提高;红外光谱结果表明,协同作用的起源是双金属催化双核消除,而不是团簇催化。2003年,该研究团队又提出了第2个双金属催化双核消除的例子,其在采用均相催化环戊二烯氢甲酰化反应制得环戊二醛的实验中发现,同时使用羰基铑和羰基锰配合物时,醛的产率有非常显著的增加;且对其进行的原位光谱和动力学测试结果表明,双金属催化双核消除四羰基铑的氢化锰配合物对醛的形成有显著贡献[42]。
刘倩等[43]用MOF-5晶体作催化剂载体,将Rh、Ni元素负载在MOF-5上合成了Rh-Ni@MOF-5双金属催化剂,分别采用1-己烯、1-辛烯和1-癸烯3种烯烃的氢甲酰化反应评价了催化剂的性能。结果表明,3种烯烃的转化率均在70%以上,其中1-己烯的转化率最高(90%),随着碳链长度的增加,转化率呈递减趋势;与单金属Rh@MOF-5相比,Rh-Ni@MOF-5双金属的转化率提高了20%左右,且产物选择性更高,表明Ni的加入有利于烯烃与催化剂的有效接触,Ni与Rh的相互促进作用提升了对烯烃的催化效率,且催化剂重复使用5次后仍可保持较高的催化活性和结构稳定性;此外,催化剂可通过离心分离等简单过滤手段进行回收,解决了回收分离难的问题。
Kartashova等[44]制备了一种由P-N配体连接的阳离子Rh-Ru配合物,并在1-辛烯的氢甲酰化反应中评价了其催化性能。结果表明,双金属络合物和单金属铑膦配合物在1-辛烯氢甲酰化中获得了相似结果,即高转化率(> 90%)、高醛产率(> 86%)以及低l/b比(< 3);进一步研究发现,Ru对烯烃的异构化和氢化影响很大,单金属Ru配合物的异构化率可达62%~80%,加入膦配体后可降低至0%~5%。并且无论是Ru、Rh单金属催化体系还是双金属催化体系,膦配体(PPh2Py)均可以在很大程度上抑制烯烃异构化。核磁共振实验和催化实验表明,Rh-Ru配合物在合成气压力下不稳定,产生了几种Rh和Ru单金属物种。但从综合性能及经济成本来看,Rh-Ru配合物具有一定的研究意义。
Chen等[45]合成了钛酸盐纳米管(TNTs)负载的Rh、Au以及Rh-Au催化剂,并考察了Au的质量分数、光还原和乙醇还原工艺以及金属负载顺序等因素对催化剂在醋酸乙烯氢甲酰化反应中催化性能的影响。结果显示,Au的加入使CO在催化剂表面的吸附量显著提高,提供了更多的反馈电子,从而增加了更多的活性位点;其中先用光还原法将Rh负载到TNTs上得到Rh/TNTs,再用乙醇还原法将Au负载到Rh/TNTs上,制得的Au0.52/Rh0.32/TNTs-12催化剂平均粒径为13 nm,该催化剂在醋酸乙烯氢甲酰化反应中表现出最高的催化活性,醛选择性达到88.67%,相比Rh/TNTs提高了28.38%。催化剂表征结果显示(图8),Rh和Au物种高度分散在载体上。但随后的循环利用结果表明,该催化剂在重复使用3次后,由于Rh和Au原子的损失导致催化效果显著下降,这可能是由于在反应过程中,金属物种与底物或合成气相互作用,使得金属纳米粒子从载体上脱落并溶解在反应体系中,从而导致催化活性下降。

图8 Au0.52/Rh0.32/TNTs-12的HAADF-STEM 图(a)及A标记区域中O、Rh、Ti 和Au的EDX映射(b) [45]Fig.8 (a) HAADF-STEM map of Au0.52/Rh0.32/TNTs-12 and (b)EDX mapping of O, Rh, Ti and Au in A labeled region [45]
综上,Rh系双金属催化剂的研究重点在于Rh-Rh双核及Rh-Fe双核催化剂。前者需要配体连接以发挥催化剂的最大活性,配体与两个Rh之间的螯合作用不仅使得催化剂更加稳定,避免了膦配体的过量使用,而且催化剂体系中没有Rh-Rh键,制备过程更加简单;但在反应过程中,易发生Rh-配体键断裂的情况,且制备过程可能会消耗大量的Rh。Rh-Fe双金属催化剂由于经济成本低而备受关注,该催化体系在合适的催化条件下对高碳烯烃的氢甲酰化催化性能与工业上应用的铑膦催化剂相当,但此类催化剂需要较高的反应温度,且存在诱导期,尽管如此,Rh-Fe双金属催化剂仍然是很有发展前景的催化体系。
为更直观反映不同催化体系的性能,将各双金属催化体系反应特点汇总如表1所示。

表1 各双金属催化剂体系对比Table 1 Comparison of bimetallic catalyst systems
3 结语与展望
为了提升氢甲酰化反应催化剂的性能及降低催化剂成本,研究者们开展了基于Co、 Rh的双金属催化剂研究。本文综述了用于氢甲酰化反应的Co系和Rh系双金属催化剂的研究进展,并对其催化性能进行了分析对比。结果表明,在Co系双金属催化剂中,Co-Rh双金属催化剂由于双金属之间的协同作用增加了催化体系的活性及稳定性,烯烃转化率与产物醛的选择性均在80%以上,且更适用于低碳烯烃的氢甲酰化反应;同时,负载型双金属催化剂解决了均相反应分离难的问题。Rh系双金属催化剂中的Rh-Rh双核催化剂由于Rh与配体之间的螯合作用使得催化剂更加稳定,可减少膦配体的用量,且由于没有Rh-Rh键,制备过程更简单;但在氢甲酰化反应过程中由于Rh-配体键易断裂,需要消耗更多的Rh,成本相对较高。Rh-Fe双金属催化剂具有更高的烯烃转化率(> 95%)、更高的醛产率(> 90%)、更长的使用寿命以及更低的成本等优势;存在的不足是反应温度偏高(70~100 °C)。Rh-Co系双金属催化剂不局限于长链或短链烯烃的应用,适用范围更广,是一种很有前途的无配体非均相氢甲酰化催化剂。
目前,氢甲酰化催化剂的研究主要朝着易分离、高活性、长寿命和低成本几个方向发展,在Rh价格昂贵且资源稀缺的情况下,双金属催化剂通过加入第2种金属来减少Rh的用量,同时又能满足高活性、易分离的要求,具有很大的发展前景;此外,双金属催化剂的多相化,如负载至SiO2、MOF、碳材料以及Fe3O4上,或采用两相溶剂等也是未来双金属催化剂的重要研究方向。
