深度学习辅助药物发现的研究进展
2022-03-06戴青青余俊霖李国菠
戴青青,余俊霖,李国菠
(四川大学华西药学院药物化学系,四川 成都 610041)
人工智能(artificial intelligence,AI)概念始于1956年,经过半个世纪的曲折探索,于2011年进入蓬勃发展时期,目前已成为一门新的技术科学,推动人类进入智能时代。深度学习(deep learning,DL),又称为深度神经网络,是AI领域中一个热门研究方向,其通过对样本数据进行多层次的非线性信息处理和抽象,挖掘内在规律,用于解决特征学习、分类和模式识别等问题。当前主流的DL模型包括卷积神经网络(convolutional neural network,CNN)、循环神经网络(recurrent neural network,RNN)和图神经网络(graph neural network,GNN)等,以及这些模型的变体,如残差卷积网络模型(deep residual network, ResNet)、变分自编码器(variational autoencoder,VAE)、对抗自编码器(adversarial autoencoder, AAE)、生成对抗网络模型(generative adversarial network,GAN)以及信息传递网络模型(message passing neural network,MPNN)等,这些DL模型在图像识别、语音识别、机器翻译、人机对弈、无人驾驶等方面已取得了前所未有的成效,深刻地改变着人们的生产生活方式[1-2]。
同时,DL技术在医学、药学、生命科学等领域也逐渐崭露头角。例如,2018年Waller团队通过DL网络对1 240万个单步反应进行化学转化规则提取,再利用3种不同的神经网络与蒙特卡洛树搜索结合形成的新算法,实现了化合物合成路线的高效设计[3]。随后, Jensen和Jamison团队又报道了一种集成合成路线设计和自动化合成的平台,并完成了15个小分子药物的自动化合成,进一步推动了该领域的发展[4]。近期,Hassabis团队报道了新蛋白结构预测工具AlphaFold2,通过将蛋白结构的物理和生物知识整合到DL方法中,极大程度提高了蛋白结构预测的准确性[5];与此同时,Baker团队也报道了新蛋白结构预测工具RoseTTAFold[6],其采用了注意力机制使整个DL能够同时学习到蛋白一级/二级/三级结构不同维度的信息,预测准确率与AlphaFold2不相上下。此外,近几年还发展了若干DL方法用于药物-靶标相互作用预测、药物靶标预测、药物从头设计、药物性质[主要包括吸收、分布、代谢、排泄、毒性(ADMET)]的预测,从而服务于创新药物研发的多个重要环节。这些工具或将改变创新药物研发进程,提升药物研发效率。鉴于此,本文聚焦DL在创新药物发现中的发展和应用,综述具有代表性的DL案例和研究思路,总结其应用特点、面临的问题及可能的解决策略,期望为DL在药物发现领域的发展提供借鉴和思考。
1 基于深度学习的蛋白结构预测
蛋白质三维结构是药物靶标功能研究与药物设计的重要基础,如何快速高效获得准确的蛋白质结构是需要解决的科学问题。早期阶段,研究人员基于统计的蛋白质进化信息,并采用传统的机器学习方法(如蒙特卡罗方法、支持向量机等)和全连接神经网络(fully-connected neural network,FNN)模型实现蛋白质三维结构的预测。例如,Bohr等[7]和Fariselli等[8]使用目标蛋白一级序列、同源蛋白序列以及关联突变等数据来训练FNN模型,实现对蛋白质主链结构的预测,但距离实现蛋白质三维结构精准预测仍有较大差距。
随着蛋白结构数据的不断增加和DL技术的迅猛发展,更复杂的深度网络模型和更丰富的蛋白质序列信息被应用于预测蛋白质的三维结构,突破了从蛋白质一级序列直接得到蛋白质三维结构的瓶颈,预测精度接近实验解析水平。基于DL的蛋白结构预测是研究人员一直在尝试和努力的方向,大致流程是通过序列比对得到进化相关的多序列比对(multiple sequence alignment,MSA)特征,联合蛋白序列编码作为输入,利用深度网络模型预测残基间的接触图或更具体的距离分布,以及蛋白骨架的二面角分布,然后将预测的空间结构信息作为约束条件,重构出蛋白三维结构(见图1)。例如,Hassabis团队最新报道的蛋白结构预测工具AlphaFold2,在最近的蛋白质结构预测技术评估(即The 14th Edition of Critical Assessment of Structure Prediction,CASP14)比赛中取得最佳预测名次,全局距离测试(global distance test,GDT)中位数得分达92.4,达到实验解析水平。AlphaFold2是基于注意力机制的神经网络模型,由Evoformer网络模块和结构生成模块组成,通过给定的一级序列,结合学习蛋白结构的物理和生物知识,端对端直接生成蛋白的三维结构。Baek等[6]也基于注意力机制开发了一种新的端到端蛋白结构预测工具RoseTTAFold。该工具是一种三轨网络模型,分别用逐级连接的网络来传递和处理来自蛋白一级、二级、三级结构的信息,轨道之间的多次连接让网络能够同时学习序列、残基间距离和原子坐标之间的关系。实验结果表明,RoseTTAFold不仅预测精度接近AlphaFold2,为未知结构蛋白生物学功能和机制提供一种解释,而且还能直接根据序列信息快速构建出准确的蛋白-蛋白复合物结构。在所需计算资源和计算时间方面,RoseTTAFold较AlphaFold2也显示出一定的优势,除去序列比对和模版搜索所用时间,其仅需1个图形处理器(graphic processing unit,GPU)就能在10 min之内生成蛋白3D主链结构。另外, Rahman等[9]在ResNet模型的基础上进行了改进,提出了一种用来预测蛋白质残基间距离的DL模型,相对比以上方法使用更少的蛋白特征,包括2种共同进化特征和3种非进化特征,实现对蛋白质残基间真实距离的高精度预测,与最先进的同类方法相比,局部距离差测试平均分数提高了10%以上,为蛋白质结构预测提供了一种新的参考。
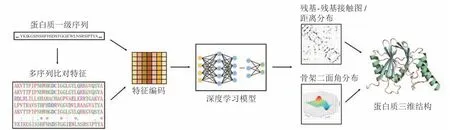
图1 基于深度学习的蛋白质三维结构预测流程Figure 1 The general process of deep learning-based 3D protein structure prediction
此外,Yang等[10]首次提出利用GAN模型预测蛋白质残基-残基接触图,并在基准测试集上表现出不错的预测效果。该模型被命名为GANcon,GANcon通过对抗性学习策略训练生成模型和判别模型,最终能够生成接近真实数据分布的接触图。其中,生成模型采用编码器-解码器框架从多种蛋白质序列特征中捕捉潜在的残基间接触信息,从而生成仿真的残基接触图;判别模型则选用基于残基块的CNN,以生成的或真实的接触图——蛋白质序列特征样本作为输入,识别生成的接触图与真实接触图之间的差异,驱动生成模型生成更准确的接触图。他们还引入了一种新的对称焦点损失函数,用来解决接触图内数据不平衡问题。但GANcon在训练过程中的不稳定性以及输入特征的选择等方面仍有改进空间。
2 基于深度学习的药物-靶标相互作用预测
药物-靶标相互作用(drug target interaction,DTI)是药物发现的重要基础,准确有效的DTI预测能极大地助力药物研发,加速先导或苗头化合物发现。近几年,基于DL预测DTI的方法陆续被报道,其一般工作流程如图2所示,研究人员针对药物和靶标的结构以及理化性质构建各具特色的描述符,并采用不同的DL网络模型,学习DTI规律,最终预测出DTI的可能性或者相互作用强度。

图2 基于深度学习的药物靶标相互作用预测一般流程Figure 2 The general process of deep learning-based drug-target interaction prediction
早期研究人员倾向于使用简单直接的输入数据和结构单一的网络框架。例如采用药物结构信息和靶标的序列信息,通过基础版本的RNN、CNN等模型学习相互作用特征[11-12],但预测结果并不理想。研究人员分析发现只是纯粹地使用药物-靶标相关信息套用DL模型不能从根本上解决问题,需在DL和药物发现的双重理论指导下,根据药物、靶标的各种性质合理构建输入描述符,同时搭建适应药物-靶标体系的神经网络框架,才能有效提高模型的预测能力和结果可靠性。在此基础上,发展出了一系列基于格点、基于图结构以及新算法的DL网络,并合理引入注意力机制等算法增强模型的可解释性。
基于格点的特征构建方法蕴含更加丰富的空间信息,比较适应于DTI预测体系。由此方法构建的特征可以视作一幅三维图片,可配合使用三维CNN模型进行训练、学习,但存在参数量大、计算成本高等问题。Li等[13]借鉴ShuffleNet、Xception等轻量级三维CNN模型[14]并构建了DeepAtom模型,用于预测药物-靶标亲和力。除了具备三维CNN模型的各种优势,DeepAtom模型同时通过深度可分离卷积解决了三维CNN模型参数过多的问题,并利用多个小的卷积核代替单个大卷积核,达到减少参数的同时增加网络复杂度的目的。该模型在PDBbind(2016版)核心测试集预测的皮尔森相关系数达0.831,表现出较强的预测能力。
Zheng等[15]对DTI预测有着不同理解,他们将DTI预测抽象成虚拟问答(visual question answering,VQA)问题,采用药物SMILES和靶标残基距离矩阵作为输入,并基于CNN与RNN模型构建了DrugVQA模型,同时引入了注意力机制以增加模型的可理解性。经过训练及超参数优化,DrugVQA模型最终在数据库DUD-E上表现出不凡的预测能力,受试者工作特征曲线下面积(area under the receiver operating characteristic curve,ROC-AUC)达到0.972。
GNN模型在此领域也备受关注,Cho等[16]采用了一种特殊的GNN模型,提出了InteractionNet框架,用于预测药物-靶标之间的结合常数。InteractionNet模型是一种非常规的GNN模型,在对药物-靶标体系建模时除了考虑共价键外,还考虑了非共价作用,最后基于PDBbind数据集采用20折交叉方法进行验证,其均方根误差(root mean square error,RMSE)为1.321,优 于PoteintialNet模型(RMSE为1.343)。
Zeng等[17]认为通过拼接药物和靶标的特征向量来表征二者的相互作用,并不能准确描述二者复杂作用体系,需要某种特殊的算法或网络来解决。据此,他们提出了一种多注意力模块MATT_DTI,首先通过相对自注意模块提取药物的化合物原子间联系,用CNN模块分别学习药物和靶标的隐含信息,最后通过多头注意力模块和全连接层提取相互作用信息并给出预测结果。该方法在KIBA和Davis数据集上表现良好,均比同类模型有更好的预测效果,如用KIBA数据集进行测试,MATT_DTI模型平均标准误差(mean squared error,MSE)在0.15左右,低于其他基准模型的MSE指标。Sajadi等[18]以药物指纹矩阵和药物-靶标矩阵为输入,构建了一个无监督去噪自编码器(denoising autoencoder,DAE)模型,并将其命名为AutoDTI++。该方法在G蛋白偶联受体(G protein-coupled receptor,GPCR)数据集上预测随机药物靶点对时,ROCAUC值达0.85,与类似算法的模型测试结果相比有明显提升。
3 基于深度学习的药物靶标预测
药物靶标预测可以帮助研究人员确定已知药物或活性分子的潜在靶标,从而有助于实现老药新用、药物重定位、毒性预测等。上述DTI预测方法也可以用于药物靶标预测。除此之外,基于异质网络等DL方法也被用于药物靶标预测,其特点在于利用药物-疾病信息、靶标-靶标信息、药物-靶标信息等多维度信息(见图3)作为网络输入特征,将其进一步转化为一组DL模型可处理的特征矩阵,实现对药物靶标的预测。

图3 基于机器学习——异质网络的药物靶标预测方法一般流程Figure 3 The general process of machine learning (heterogeneous network)-based target prediction
自编码器(autoencoder,AE)及其变体,如DAE等在基于异质网络的靶标预测方法中较为主流,研究人员通过收集药物、靶标相关的各种信息,构建异质网络,利用各种AE变体进行学习,最终分析和预测药物的潜在靶标。Zeng等[19]收集了药物-疾病、药物-不良反应、药物-靶标、药物-药物相关信息,以此构建异质网络,从中提取药物与靶标之间的关系,使用随机游走算法计算得到概率共生矩 阵(probabilistic co-occurrence matrix,PCO),再计算正点互信息矩阵(positive pointwise mutual information,PPMI)来表征异质网络整体结构,用于训练DL网络模型,由此发展了deepDR模型。该模型在基准模型上,deepDR预测效果更佳,ROCAUC达0.908。后来,他们又进一步做出了改进[20],设计了一个新的模型(deepDTnet),该模型在输入和框架方面都进行了优化,丰富了异质网络所蕴含的信息,加入了更多靶标相关信息,如靶标-靶标相似性、靶标-疾病信息,同时保留PCO矩阵和PPMI矩阵的表征方式,采用多层DAE学习异质网络的隐含信息。与deepDR相比,deepDTnet具有更强的预测能力,ROC-AUC达0.963。也有研究人员通过将AE和其他网络模型结合,尝试发展了新的网络模型。如Peng等[21]提出了DTI-CNN模型,特点在于使用Jaccard相似性系数结合重启随机游走算法(random walk with restart,RWR)来提取药物特征和靶标特征,且经过DAE层后添加了CNN模块来预测最终结果,训练后ROC-AUC达0.9416,与deepDTnet效果相当。
除了AE及其变体外,其他模型在药物靶标预测方面也展现出不俗的预测效果。Manoochehri等[22]利用更简单的输入(仅考虑药物-药物相似性和靶标-靶标相似性信息)和FNN模型进行学习预测,但将更多的精力放在输入数据的处理上,提出了独特的特征提取和构建方法。他们利用异质网络的拓扑结构来预测药物的未知靶标,通过药物-药物相似性和靶标-靶标相似性信息把药物-靶标异质网络抽象成半二部图,并从中提取出多个封闭子图,然后采用Weisfeiler-Lehman算法对每个子图中的节点进行排序标记,以表征药物-靶标对的拓扑结构。最后使用这种特殊的输入来训练FNN模型,同时进行了10折交叉验证。结果显示,该方法比 BLMNII、CMF、HNM等同类模型预测能力更强。此外,GNN模型也被用来处理这些异质网络,进行药物靶标的预测。Huang等[23]提出了SkipGNN模型,并认为异质网络中直接相连的2个节点并不一定有很强的相似性,反而是间接的或跳跃的节点间的相似性可能更加必要。根据这种思想,他们以药物-药物、靶标-靶标、药物-靶标、基因-疾病相关信息构建了异质网络,从中提取跳跃相似性信息并构建跳跃相互作用图,同时结合原始图输入至GNN模型中,最后经由解码器输出药物与靶标相互作用概率。实验结果表明SkipGNN模型优于其他模型,如DeepWalk、图卷积神经网络(graph convolutional neural network,GCN)和node2vec模型等。
4 基于深度学习的合成路线设计
药物研发离不开合成路线设计,设计高效的合成路线可大幅度降低药物研发成本、缩短生产周期、提高药物研发效率。传统的计算机辅助合成路线设计的方法主要是基于大量 “专家”规则和逆合成分析方法来规划合成路线,但其存在设计速度较慢、设计的合成路线往往不太合理等问题[24]。随着DL算法在化合物性质预测和生物活性预测等领域中展现出巨大的潜力,其也逐渐被应用于合成路线的设计并取得了一定的进展。
Waller团队于2018年报道了一种AI工具3N-MCTS,通过使用3种不同的深度神经网络(分别是拓展策略网络、筛选网络和展示策略网络)和蒙特卡洛树搜索算法来设计目标化合物的合成路线[3]。他们首先利用拓展策略网络对目标分子进行逆向化学转换,搜索当前节点可能的变换路径,然后使用筛选网络分析判断反应是否可行,过滤不合理的反应路线,最后通过展示策略网络多次随机采样对搜索节点进行评价打分。研究人员利用来自Reaxys数据库的1 240万条反应数据训练这些网络,学习化学转化规则。与其他方法相比,3N-MCTS在合成路线的搜索速度、质量等方面均有显著提升,能在短时间内生成数百个化合物的合成路线,且双盲实验结果表明3N-MCTS预测分子合成路线水平接近合成化学家水平。这种方法的优势体现在无需专家自定义规则,DL模型就可以学习到已知反应所蕴含的转化规则,然后根据学习到的规则快速选择出最佳合成路线。
随后,Coley等[4]推出了一个基于AI的自动化合成平台,首先利用前馈神经网络生成目标分子的合成路线,然后机器人根据合成方案执行一系列具体的制备过程,实现自动化合成。研究人员使用Reaxys和USPTO数据库中的反应数据训练网络模型,学习反应转换规则,为目标化合物设计出可行的合成路线,包括给出反应条件,同时根据合成路线中的反应类型是否容易实现以及中间产物是否多样化等条件进一步筛选得到最优合成路线。最后,他们通过该平台成功完成了15种小分子药物合成路线设计并实现了自动化合成。同时,基于DL的序列到序列(sequence-to-sequence,seq2seq)模型(如Transformer模型等)的发展给不依赖模版的逆合成预测任务提供了一种新的解决思路(见图4):可将该任务看成自然语言处理(natural language processing,NLP)领域内机器翻译任务,输入目标分子的SMILES序列,不依赖反应规则,就能输出对应单步的反应物SMILES序列。

图4 基于序列到序列模型进行合成路线预测Figure 4 Prediction of synthetic route based on the seq2seq model
Liu等[25]率先将seq2seq模型应用到逆合成预测任务中,使用的seq2seq模型是基于RNN的编码器-解码器结构,并在包含5万个专利反应的数据集上训练,并初步达到了与基于规则的基准方法效果相当的水平。该方法在一定程度上突破了专家规则的限制,并表现出良好可扩展性的优势。随后seq2seq模型经过发展,得到了较为流行基于注意力机制的Transformer模型。Zheng等[26]开发了一种无模板的自校正逆合成路线预测工具SCROP,通过使用基于多头注意力机制的Transformer网络模型预测逆合成路线,同时引入了基于Transformer的语法校正器,对预测模型产生的不合理候选前体分子SMILES进行修正。SCROP在基准数据集上预测准确率达59%,比基于模板的方法提高了6%;同时实验结果表明语法校正器的加入提高了模型预测质量,使无效的候选前体分子比例从12.1%降至0.7%。此外,Guo等[27]结合Transformer模型和贝叶斯推理算法进行逆向合成预测。他们将该任务视为组合优化问题,即在所有可用的反应物组合中找到一组最佳的反应物对,用来合成目标产物。他们首先通过训练好的Molecular Transformer模型对给定反应物组合进行高精度正向预测,然后基于贝叶斯定理将正向预测模型反演为逆向合成模型,同时使用蒙特卡罗搜索算法探索得到最佳的反应物组合。正向和逆向预测模型的组合提高了合成路线的可行性,同时改善了逆合成问题的不适定性。
这类序列模型一般利用分子的SMILES字符串作为输入,未能有效刻画出分子中各原子间复杂关系。为此,Shi等[28]提出了一种基于图神经网络的无模版逆合成预测框架G2G(graph to graph framework),利用图表征分子,将任务转化为图到图的翻译问题,即将目标分子图转化为一组反应物分子图。研究人员首先基于GCN识别目标分子的反应中心,将目标分子拆分为一组合成子。然后,通过图VAE将每个合成子转换为最终的反应物子图。实验结果表明G2G在Top-1准确率指标上明显优于其他无模版的基准模型(如seq2seq模型、transformer模型等),并与最先进的基于模板的方法相比水平相当,如条件图逻辑网络(conditional graph logic network,GLN)模型。
5 基于深度学习的从头药物分子设计
近年来在从头药物分子设计领域,DL方法因部分解决了传统方法的组合爆炸、多目标优化等问题而受到越来越多的关注。许多相关研究都证明了DL方法在从头药物分子设计的可行性,目前关于DL在这方面的应用已经被总结报道[29-31],在此笔者将对最新的研究进展进行进一步介绍。Born等[32]构建了一种混合的VAE模型,用来生成具有抗癌药物特性的候选分子。值得注意的是,他们不仅使用分子SMILES作为输入,还首次加入疾病相关的基因表达数据,同时使用抗癌药物敏感性预测模型作为奖励函数。混合的VAE模型由2个并列的VAE组成,一个用于接收小分子SMILES以学习其语法规则,另一个VAE用于接收基因表达数据以学习其特征表示,然后将这2个VAE编码器的输出结果一并输入到同一解码器,生成新分子,最后用抗癌药物敏感性预测模型预测生成分子对靶细胞的活性值。应用在4种不同癌症类型的实例表明,该模型能够针对特定疾病生成具有较强抑制效果的分子,且生成的分子在结构、可合成性以及溶解性等方面均与现有药物相似。然而,VAE也存在一定局限,它只会最大限度地“模仿”训练数据,尽可能生成与训练数据在结构上相似的分子,因此生成分子的结构新颖性较低。
AAE在VAE基础上增加了判别模型,对采样分子和真实样本进行区分,基于对抗的思想训练生成模型和判别模型,扩展了分子的生成空间,一定程度上弥补了VAE在生成分子时结构新颖性方面的缺陷。Polykovskiy等[33]构建了一种新的AAE模型,即条件AAE,其能够基于指定条件(如药物分子的靶标特异性、溶解性、可合成性等)生成相应的分子。其中,基于长短时记忆网络(long short term memory,LSTM)分别构建编码器和解码器,同时使用多层的FNN作为判别模型,用来判断采样分子是否符合真实数据分布以及是否具备所需的理化性质,并基于半监督学习方法优化模型。他们利用该模型成功发现了一种新型的Janus激酶3(Janus kinase 3,JAK3)抑制剂。
Bagal等[34]受生成式预训练新型神经网络模型(generative pre-training transformer,GPT)Transformer在生成文本任务中取得突破性进展的启发,基于GPT构建了一个新的生成模型MolGPT,能够根据给定条件(输入SMILES字符串、脂水分配系数、可合成性分数以及拓扑极性表面积等目标属性值)生成具有所需骨架和理想特性的分子。MolGPT由多个堆叠的解码器模块组成,每个解码器包含一层掩码自注意力层和多层全连接网络,能够捕获SMILES字符串中字符间远距离依赖关系。与VAE、AAE等其他DL模型相比,MolGPT在生成分子的有效性、独特性以及新颖性方面表现较好,打分分别为0.981、0.998和1.0。
Goel等[35]结合RNN和强化学习,提出了一个分子生成模型MoleGuLAR,其能够对分子的类药性、结合亲和力等方面进行多目标优化。尤其是,他们提出一种新的交替奖励策略,奖励函数随着生成不同分子的过程中在动态地改变,使得模型能够交替探索不同的化学区间,采样得到更加合理的分子。区别于以往大多数DL模型只能生成一维或二维的分子,Li等[36]将DL与基于结构的从头药物设计策略相结合,发展了一种新的从头分子生成模型DeepLigBuilder,其能够直接生成具有高结合亲和力类药分子的三维结构。DeepLigBuilder首先利用一种图生成模型即配体神经网络(ligand neural network,L-Net)实现生成类药分子的三维结构,然后结合蒙特卡洛树搜索方法将靶标的结构信息引入到模型中,在靶标活性位点搜索、优化分子的结合构象,从而得到具有高结合亲和力的新分子。通过将其应用于严重急性呼吸综合征冠状病毒2 (severe acute respiratory syndrome coronavirus 2,SARSCoV-2)抑制剂的从头设计,他们得到了3种新型具有高预测结合亲和力且与已知抑制剂结构类似的SARS-CoV-2潜在抑制剂,证明了DeepLigBuilder在从头药物设计以及先导物优化方面的实用性。
为了解决DL在小规模训练数据集上表现较差等问题,Krishnan等[37]设计了一套基于RNN的生成模型和迁移学习的药物从头设计流程,生成的分子不仅具有所需类药特性,同时还具有靶标特异性。他们首先利用ChEMBL数据库中的活性分子SMILES数据预先训练RNN生成模型,以学习SMILES语法规则;然后,通过对接得到具有靶标选择性的分子并进行迁移学习,生成作用于特定靶标的分子;同时,再建立另一个基于RNN的预测模型,作为奖励函数评价生成的分子与靶标的结合亲和力。另外,Moret等[38]将RNN生成模型与数据增强、温度采样和迁移学习这3种优化方法结合起来,也能够在具有少量数据情况下生成所需特性的新分子。
6 基于深度学习的药物吸收、分布、代谢、排泄和毒性预测
药物的ADMET性质研究对于药物研发也是至关重要的。据统计,将近50%的候选药物在临床试验阶段因ADMET性质不符要求而宣告失败。因此,在早期药物发现和药物设计阶段,研究人员应提前对药物分子的ADMET性质进行预测评估,以降低后续临床试验失败的风险。相较于耗时耗力的实验方法,精确可靠的ADMET预测方法能极大地缩短时间花费、减少实验成本,提高候选药物的筛选效率,基于DL的ADMET预测方法则恰逢其会,并逐渐成为预测药物ADMET性质的重要手段。
近几年来,利用DL方法来预测小分子性质已经较为普遍,其中基于GNN模型的方法受到了学界的广泛认可,预测结果相较其他DL方法更为可靠。2018年,Wu等[39]基于DeepChem平台构建了一个用于分子性质预测的DL框架,称为MoleculeNet。他们通过这个框架为同行提供了一个基准,可以用于比较各种不同模型的效果和可靠程度。该框架涵盖了不同的数据集拆分方法,包括基于骨架、随机拆分等;以及不同的特征构建方法,处理为ECFP、图结构等;和不同的网络模型,例 如GCN、MPNN、weave、随 机 森 林(random forest,RF)、核岭回归(Kernel ridge regression,KRR)等;并针对各种ADMET性质相关的数据库(如QM8、Clintox、Lipophilicity、BBBP等)进行训练和测试。通过一系列基准测试,他们发现在应用量子力学性质、物理化学性质、生理学性质相关的数据集时,最佳的GNN模型比最佳的传统模型更为有效,如应用QM8数据集训练模型并预测小分子量子力学性质时,以平均绝对误差(mean absolute error,MAE)为评价指标,表现最佳的传统模型是KRR模型,该模型MAE达0.015,而基于GNN的网络模型中表现最佳的是MPNN模型,其测试结果MAE为0.014 3,误差低于KRR模型测试结果。随后研究人员从不同角度出发,建立了一系列各具特色的GNN模型。 Feinberg等[40]构建了一种新型GNN网络模型PotentialNet,其核心思想是在更新原子状态过程中考虑距离因素,比常用的邻接矩阵更能描述药物分子结构。该方法相较于传统的机器学习方法和一些常见的GNN模型性能更佳,仍以QM8数据集进行测试,在基于此数据集预测小分子量子化学性质任务中,MPNN在测试集上MAE达0.013 9,而PotenialNet则提升明显,MAE在0.011 8左右。后续研究中,他们又进一步在PotentailNet模型基础上进行了改进,设计出多任务PotentialNet模型,同时采用31项ADMET性质进行训练,最终同时预测这31项性质[41],例如电压门控钾离子通道(human ether-à-go-go-related gene encoded potassium ion channel,hERG)抑制性、人肝细胞清除率、半衰期、脂溶性等,并与RF模型进行了比较。对于绝大部分性质而言,多任务PotentialNet模型预测的相关系数(R2)与RF模型相比都有不同程度的提高,例如以时序拆分方法拆分数据集时,多任务PotentialNet模型较RF模型,在31项性质预测中R2平均高出64%。
Yang等[42]则开发了一种有向信息传递网络(directed message passing neural network,D-MPNN),与往常的GNN模型做法不同,在表征药物分子结构时,他们将原子间的键考虑为有方向的边,而非常规的无向的边,且通过边的方向来对原子的状态进行更新,减少了无效冗余的原子状态更新。预测结果表明,在所有数据集上D-MPNN都比RF模型、FNN模型等性能更好或者相当;例如,在血脑屏障透过能力预测方面,D-MPNN模型的ROC-AUC高达0.925,而RF模型和FNN模型分别仅为0.788和0.899。Li等[43]提出了基于多头三联注意力机制的MPNN模型TrimNet,通过给定的邻接矩阵、边特征矩阵、节点特征矩阵,分析周围原子对当前原子的影响,从而实现高效地从图结构表征的药物分子结构中学习潜在信息,并大幅度减少模型参数数量、降低计算成本,最终在多个数据集上取得良好的预测结果,如在ClinTox数据集上ROC-AUC高达0.948。
除了GNN相关模型,研究人员也尝试了其他类型的DL模型,并获得一定成果。Kim等[44]开发了首个基于自注意力机制具有可解释性的DNN模型,用于预测药物是否存在hERG毒性。尽管只是采用了较为简单的ECFP描述符和FNN网络模型,但在测试集上ROC-AUC依旧高达0.893,较传统的定量构效关系(quantitative structure-activity relationship,QSAR)模型,有明显的改善。Wang等[45]基于概念新颖的胶囊网络模型(capsule neural network,CapsNet),并结合CNN、受限波尔兹曼机(restricted boltzmann machine,RBM)等网络模型构建了一系列衍生网络,用于预测药物hERG毒性,训练得到的最佳模型ROC-AUC达0.944。也有研究团队通过DL模型直接学习实验数据并预测给药后患者体内药物的药效学(pharmacodynamics,PD)/药动学(pharamcokinetics,PK)性质变化曲线。例如,最近Lu等[46]基于RNN模型和神经常微分方程(Neural-ODE)提出了Neural-PK/PD模型,其创新之处在于设计网络框架时,保留了PK/PD的一些基本原理,如药物的体内效应与给药剂量、体内浓度直接相关等,从而提升了PK/PD性质的预测准确度。
7 结语与展望
DL技术在药物发现多个环节中取得了惊人的预测能力,正在改变着药物研发进程,将有可能降低药物发现成本、提高药物研发效率。然而,现有DL技术仍面临着诸多挑战。首先,大多数DL技术严重依赖大量的计算资源,一定程度上限制了DL方法的发展及应用。如何在保持模型预测准确率的前提下,降低DL模型对计算资源的依赖已成为DL领域的一个研究热点[47]。其中一个主流思路是通过修剪DL模型或者改善DL模型结构以减少网络参数数量和运算量,从而降低对计算资源的需求。目前已有一些新型的轻量级DL模型被开发和应用[14],如SqueezeNet、ThiNet、ShuffleNet。其次,数据样本量、来源、质量等参差不齐,也限制了DL技术建立和优化。DL模型的训练依赖于大规模且高质量的数据样本。如何有效进行小样本学习是未来DL重要的发展方向[48],目前已有一些针对小样本学习的方法,如采用数据增强技术、迁移学习、多任务学习策略等。同时,数据集的质量也决定着DL模型预测性能的好坏。药物研发相关原始数据的提取、特征构建等方法尚存在不足,影响着高质量DL模型的发展。近年来,图神经网络的发展,蕴含更多结构信息的图被逐渐用来表征分子并应用于药物发现领域,已取得一些研究进展。此外,DL模型中超参数搜索、内部机制的不可解释性等,也一定程度上阻碍了该技术的发展。总而言之,以上DL技术面临的种种不足和挑战都在提示我们,需要更多不同背景的研究人员加入到这一领域,来提出更多精湛的DL算法,并且要充分结合传统的药物设计方法,才能逐步解决药物研发过程中各个环节的具体问题,从而能助力创新药物发现,进一步推动药物研发领域迈向智能时代。
