葡萄膜恶性黑色素瘤转移相关的非编码RNA表达谱及竞争性 内源RNA调控网络分析
2022-03-03陈晓云杨伟敏邓小茜季娴肖伟
陈晓云,杨伟敏,邓小茜,季娴,肖伟
(中山大学中山眼科中心,眼科学国家重点实验室,广东省眼科视觉科学重点实验室,广州 510060)
葡萄膜恶性黑色素瘤(uveal melanoma,UM)是成人最常见的眼内原发性恶性肿瘤,好发于高加索人和西班牙裔。初诊治疗后,约一半UM患者会最终发生远处转移[1]。由于缺乏有效的治疗手段,大多数远处转移的患者在发现转移灶后的存活时间通常少于一年。目前,学者们已经建立了几种肿瘤基因表达谱、临床特征和/或细胞遗传学标志物预测转移风险的模型[2]。最近,癌症基因组图谱(The Cancer Genome Atlas,TCGA)项目将UM分为4个分子上不同、临床上相关的亚型,这些亚型与不同的预后显著相关[3]。尽管在寻找转移的分子标志物方面已经取得了一定进展,但UM发生远处转移的生物学机制仍未完全明确[1-2]。
长链非编码RNA(long non-coding RNA,lncRNA)是长度大于200个核苷酸的转录本,不能编码蛋白质[4]。近年来研究[5]发现:lncRNA能与DNA、RNA和蛋白质等细胞大分子相互作用,参与肿瘤的发生发展。LncRNA可作为竞争性内源性RNA(competing endogenous RNA,ceRNA)吸附microRNA(miR),间接调控miR靶点mRNA的翻译和蛋白质合成[6]。通过这种调控方式,lncRNA促进胃癌[7]和胰腺癌[8]等多种恶性肿瘤的发生和转移。在人类UM组织中,已报道了几种致癌性lncRNA(例如HOXA11-AS[9],RHPN1-AS1[10]和PVT1[11])的显著高表达,体外实验证明它们能促进肿瘤细胞的增殖、迁移和侵袭,但尚无研究对lncRNA表达异常及其在UM转移中的作用进行系统性分析,而深入了解lncRNA在其中的作用机制可能对揭示UM转移的潜在机制具有重要意义。
本研究的目的是利用生物信息学方法,全面解析TCGA-UVM数据集中,lncRNA的表达与相关ceRNA网络在UM转移中的作用,为进一步实验研究提供生物信息学证据。
1 资料与方法
1.1 数据采集与处理
本研究从TCGA数据门户网站(https://tcgadata.nci.nih.gov/tcga/)获得所有UM患者(80例)的RNA测序数据(第3级)、miR测序数据(第3级)和临床数据。数据使用R/Bioconductor软件包的TCGAbiolinks程序进行预处理,并用biomaRt包参考GENCODE(GRCh38)进行基因注释,以下转录本类型定义为lncRNA:lincRNA、antisense、sense_intronic、processed_transcript、sense_over lap ping、3prime_over lap ping_ncRNA 和macro_lncRNA。本研究最终获得19 249 个mRNA、3 742个lncRNA和1 881个miR。根据随访期间是否发生远处转移,80例UM患者分为转移组和非转移组两组。
1.2 DEmRNA、DEmiR 和DElncRNA
使用R/Bioconductor包edgeR程序分析转移组与非转移组肿瘤样本中DE的DEmRNA、DElncRNA和DEmiR。首先利用edgeR程序内置的滤过功能删除在所有样品低表达RNA类型,再采用Benjamini-Hochberg方法计算错误发现率(false discovery rate,FDR)和DE倍数变化(fold change,FC),将满足FDR <0.05且log2(FC) >1.0的RNA定义为DE的RNA(即DEmRNA、DElncRNA和DEmiR),用pheatmap工具包绘制热图。热图聚类分析采用Ward.D层次聚类法,以欧式距离定义层次距离。
1.3 ceRNA 网络构建
每一个lncRNA-miR-mRNA相互作用组合定义为一个ceRNA 单元。我们首先从miRcode 和TargetScan数据库检索miR与mRNA的所有调控关系,并取这两个数据库的交集作为最终的miRmRNA调控作用单元;从miRcode数据库检索miR和lncRNA的调控关系。以上数据去除重复条目后,最终获得623622对miR-mRNA作用单元和 114 273对miR-lncRNA作用单元。根据ceRNA的作用原理,lncRNA和mRNA的表达水平应存在正相关,因此我们计算了mRNA和lncRNA表达水平的Pearson相关系数(Pearson correlation coefficient,PCC),并选择PCC在最高5%百分位以内的RNA入选构建ceRNA网络。
1.4 ceRNA 网络的拓扑分析和子网络构建
采用Cytoscape v3.6.0软件构建ceRNA网络,利用软件自带程序分析每个节点(即差异RNA)的中间中心度(betweenness centrality,BC),作为该节点在网络中的中心度指标,将所有节点的BC值排序,将BC值较高的lncRNA作为中枢性lncRNA,以这些lncRNA为核心,利用Cytoscape v3.6.0软件再次构建ceRNA子网络。
1.5 基因功能与富集分析
使用在线工具KOBAS 3.0[12](http://kobas.cbi.pku.edu.cn)中的基因富集模块对ceRNA网络中的mRNA进行基因功能与富集分析,将所有满足校正P<0.05(Corrected P Value)相关的基因本体(Gene Ontology,GO)和通路(Kyoto Encyclopedia of Genes and Genomes,KEGG)纳入进一步分析和展示,P值校正采用Benjamini-Hochberg法。
1.6 统计学处理
采用R软件(3.6.0版)分析数据,连续性数据采用均数±标准差(±s)表示。将DE的RNA按其表达水平中位数分为高表达组和低表达组,应用Kaplan-Meier生存曲线进行生存分析。P<0.05为差异有统计学意义。
2 结果
2.1 DE 的RNA 的筛选
从TCGA数据库下载和提取到80例UM患者的数据,按随访期间是否发生转移分为转移组23例(28.8%)和非转移组57例(71.2%),利用edgeR算法排除低表达RNA后,筛选得到15 135个mRNA,3 051个lncRNA和719个miR进行进一步分析。edgeR算法筛选得到转移组916个DEmRNA(上调346个,下调570个)、72个DEmiR(上调27个,下调45个)和272个DElncRNA(上调118个,下调154个;图1)。鉴于ceRNA的作用模式,lncRNA通过间接正向调控mRNA发挥生物学作用,在转移过程中表达升高的基因/mRNA更可能成为未来治疗的靶点。因此,为了分析具有致癌功能的lncRNA(onco-lncRNA)及其相关ceRNA网络,仅纳入上调的lncRNA和mRNA行后续研究。针对DE的RNA绘制热图,并使用Ward.D算法和欧几里得距离进行聚类分析(图1D,图2A)。
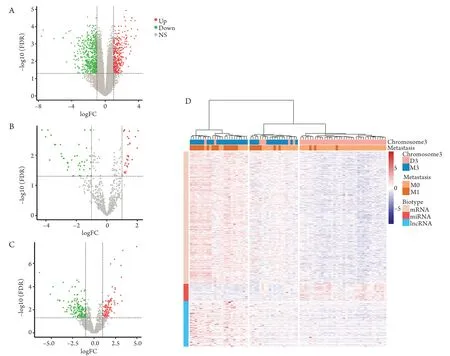
图1 火山图和热图显示UM转移组差异表达RNAFigure 1 Volcano plots and heatmap showing the differentially expressed RNAs
2.2 ceRNA 调控网络构建
为了构建与UM转移相关的ceRNA网络,首先从miR靶点数据库(即miRcode和TargetScan)获得了405个DEmiR-DEmRNA互作单元和146个DEmiRDElncRNA互作单元,并采用PCC阈值保留mRNA和lncRNA表达显著正相关的对子后,最终获得67个mRNA,7个miR(miR-221和miR-222具有相同靶点)和30个lncRNA纳入ceRNA网络,并形成616个lncRNA-miR-mRNA单元。所有DE的RNA组成了一个相互连通的网络(图2B),该网络包含103个节点和181条边。
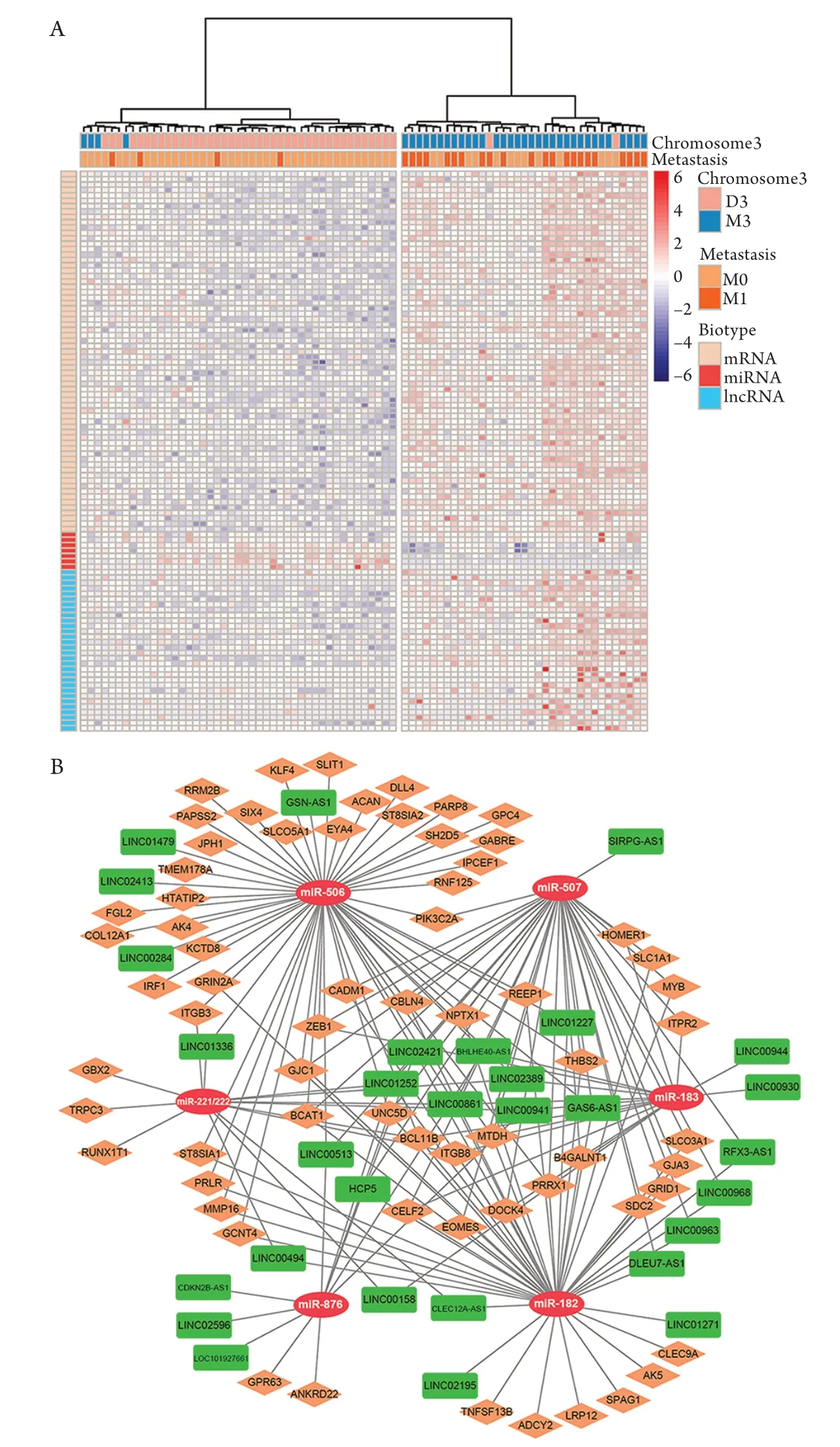
图2 热图显示构成ceRNA网络的差异表达RNA和ceRNA网络Figure 2 Heatmap of differentially expressed RNAs in the ceRNA triples and the global view of the entire ceRNA network
2.3 GO 分析与KEGG 通路分析
使用在线工具KOBAS(v3.0)对所有上调表达的mRNA进行GO生物过程富集分析和KEGG通路分析,共富集到374个GO类别,其中包括53个分子功能(molecular f unctions,MF),37个细胞成分(cellular components,CC)和284个生物过程(biological process,BP)子类,其中,最相关的生物学过程包括:多细胞生物过程、解剖结构形态发生、细胞分化调控和细胞粘附。KEGG通路分析显示有14条通路显著相关,包括细胞外基质(extracellular matrix,ECM)-受体相互作用、PI3K-Akt信号通路和R ap1信号通路,图3 显示了P值最小的前20 个GO 类别和前10 个KEGG通路。

图3 ceRNA网络中上调mRNA的基因富集分析Figure 3 Gene enrichment analysis of all upregulated mRNAs in the ceRNA network
2.4 拓扑分析和ceRNA 子网络分析
采用Cytoscape(v3.6)软件中的NetworkAnalyzer模块分析了整个ceRNA网络每个节点的拓扑特征,并计算其BC值,BC值较高的节点可能与UM转移相关性更强,从而定义为中心RNA。在ceRNA网络的103个节点中,BC值最高的前15个节点如图4A所示,包括6个lncRNA(LINC00861、LINC02421、BHLHE40-AS1、LINC01252、LINC00513和LINC02389)和3个mRNA(UNC5D、BCL11B和MTDH)然后,通过分析与核心lncRNA直接作用的一级miR,及二级mRNA来构建以该核心lncRNA为中心的ceRNA子网络(图4)。
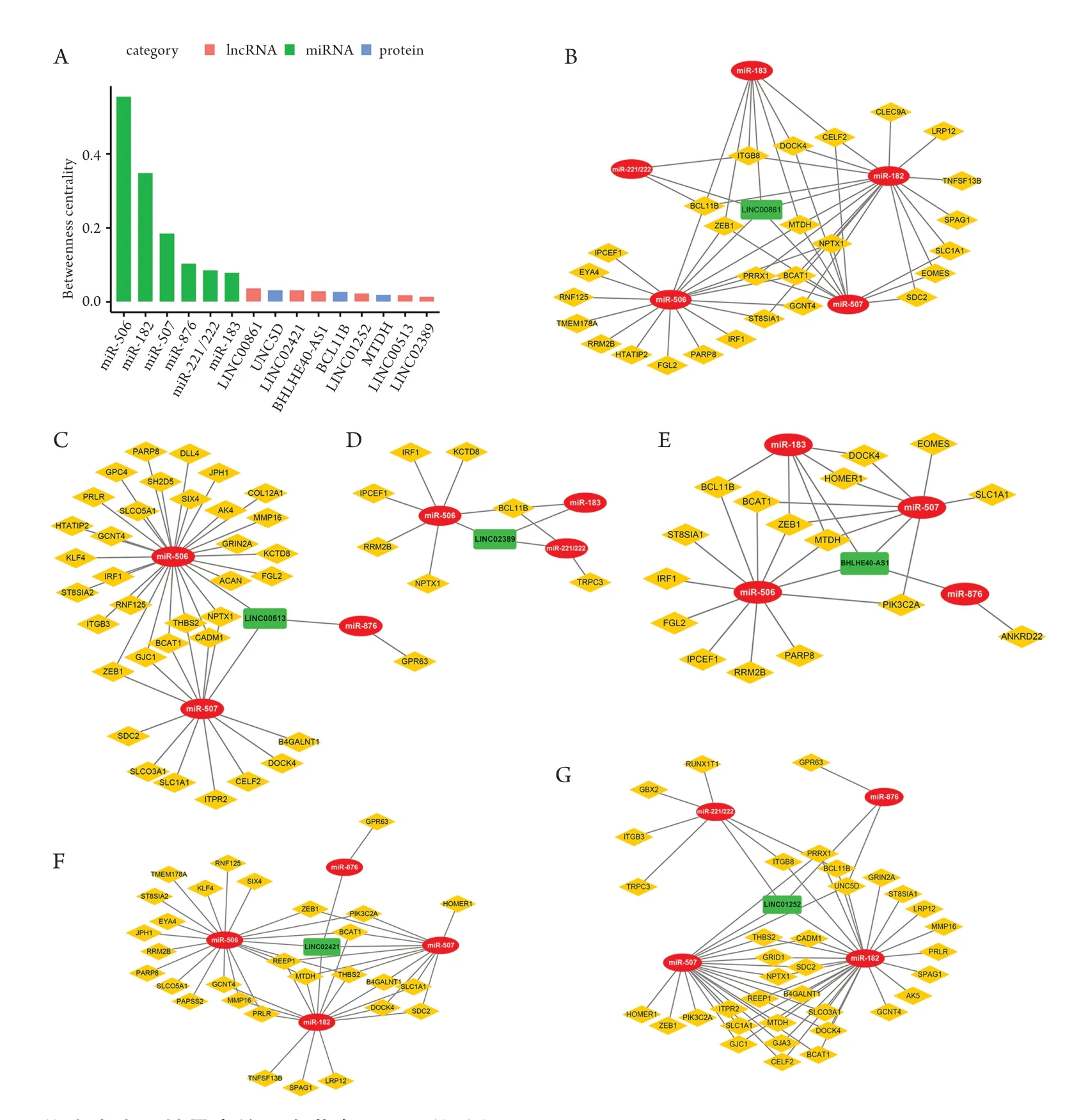
图4 ceRNA网络中介中心性最高的15个节点及子网络分析Figure 4 Top 15 nodes with the highest betweenness centrality (BC) and the subnetwork analysis of hub lncRNAs
2.5 差异RNA 表达水平与生存
Kaplan-Meier分析显示6个核心lncRNA (LINC00861,LINC02421,BHLHE40-AS1,LINC01252,LINC00513和LINC02389)、5个miR(miR-221,miR-222,miR-506,miR-507,miR-876(除了miR-182和miR-183)和3个核心mRNA(UNC5D,BCL11B和MTDH)的表达水平与总体生存率显著相关(图5,6),其中miR高表达与患者生存预后较好显著相关(图5),而lncRNA和mRNA高表达与生存预后差显著相关(图6)。

图5 Kaplan-Meier分析差异表达的miR与患者生存的关系Figure 5 Kaplan-Meier plot of the relationship between seven differently expressed microRNAs and the survival time of patients
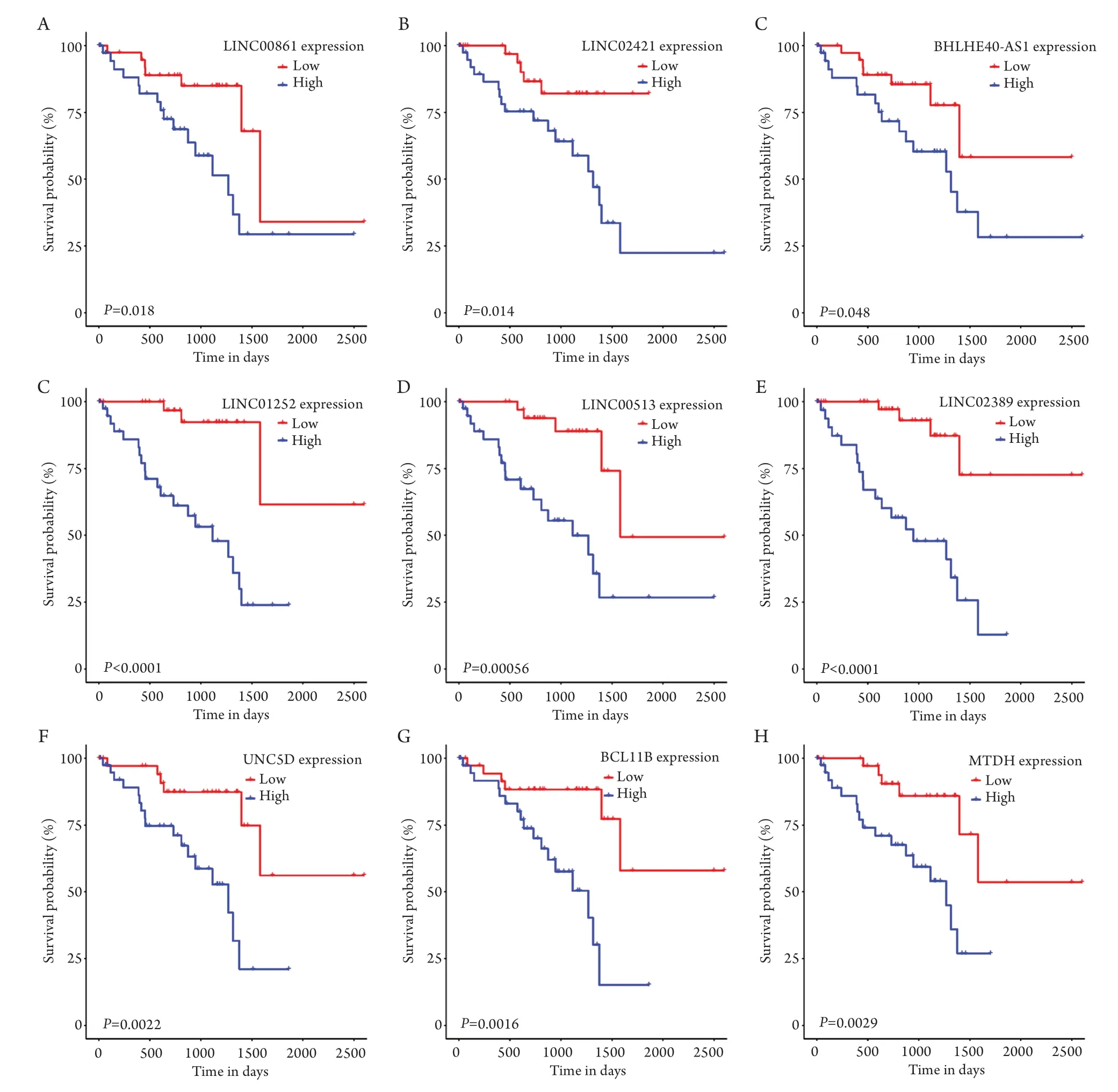
图6 Kaplan-Meier分析差异表达的核心lncRNA和核心mRNA与患者生存的关系Figure 6 Kaplan-Meier plot of the relationship between six hub lncRNAs,three hub mRNAs and the survival time of patients
3 讨论
采用成熟的生物信息学算法及高质量的TCGA-UM数据集,本研究发现多个与UM远处转移相关的编码和非编码RNA,并以lncRNA为核心建立了一个相互联系的ceRNA网络,该网络中的核心mRNA与肿瘤发生和转移的多个GO生物学过程和KEGG通路显著相关,核心lncRNA形成各自的ceRNA子网络。
LncRNAs如何在恶性肿瘤发生和转移中发挥调控功能的确切机制仍不清楚,体内和体外实验证据逐渐证明:lncRNA可以通过吸附和抑制miR的功能,间接调节癌基因的表达,从而形成了lncRNA-miR-mRNA的竞争性内源RNA的ceRNA新机制。迄今为止,许多研究发现人类恶性肿瘤中存在ceRNA调控机制[12-14],但这种机制在UM转移中是否存在,还缺少系统全面研究。本研究全面分析了TCGA-UM数据集的80个肿瘤样本的多种类型RNA表达谱,并鉴定了570个mRNA,45个miR和154个lncRNA可能参与UM转移,基于ceRNA作用原理和开源数据库(miRcode和TargetScan),最终鉴定出67个mRNA、7个miR和30个lncRNA形成了ceRNA网络,这是目前第一个关于UM转移相关ceRNA网络构建的研究。
在该ceRNA网络中,miR-182、miR-183、miR-221、miR-222、miR-506、miR-507和miR-876共7个下调的miR构成了ceRNA网络的核心。根据以往研究,其中miR-506、miR-507和miR-876的表达水平与恶性肿瘤的转移呈负相关,起抑癌基因的作用。如miR-506和miR-507可以抑制乳腺癌[15]和皮肤黑色素瘤[16]的增殖和转移。同样,miR-876可抑制头颈部鳞状细胞癌的侵袭生长[17]。这些证据与本研究在转移性UM中发现的miR-506和miR-507表达降低的结果一致。但是既往研究发现miR-221/222在肝细胞癌[18]和宫颈鳞状细胞癌[19]中发挥致癌作用,与本研究发现的转移性UM中miR-221/222的下调相矛盾,提示miR-221/222致癌和抑癌作用的发挥可能具有一定的组织和细胞特异性。
通过ceRNA网络拓扑特征分析,本研究发现6个lncRNA(LINC00861、LINC02421、BHLHE40-AS1、LINC01252、LINC00513和LINC02389)和3个mRNA(UNC5D、BCL11B和MTDH)作为ceRNA网络的中枢RNA。关于其中6种lncRNA的生物学功能的文献报道较少,有研究发现BHLHE40-AS1[20]和LINC00861[21]的表达水平升高与乳腺癌的侵袭和肝细胞癌的早期复发有关,提示它们的致癌作用,与本研究的结果一致,但对于其他4种lncRNA(LINC02421、LINC01252、LINC00513和LINC02389)的表达水平与肿瘤发生发展的关系尚无足够证据,它们是否与UM细胞的高度侵袭相关是将来实验研究的有趣方向。
与核心lncRNA 的研究相比,3 个核心mRNA(UNC5D、BCL11B和MTDH)在恶性肿瘤中的作用研究较多。有研究表明它们与多种癌症的高转移风险相关。如MTDH既可以促进多发性骨髓瘤细胞增殖,又可以抑制其凋亡[22],在肾癌[23]和结直肠癌[24]中,MTDH的高表达与远处转移风险呈正相关。在皮肤黑色素瘤中,基因敲除MTDH可显著抑制裸鼠黑色素瘤生长,也提示它在黑色素瘤中具有致癌作用,MTDH也可能成为治疗皮肤黑色素瘤的潜在靶点[25]。BLC11B也是一种癌基因,在T细胞急性淋巴细胞白血病[26]和神经胶质瘤[27]中表达上调。这些既往报道的数据均与本研究的结果一致,但它们是否同样参与了细胞增殖、迁移等生物学过程,还需要体内和体外实验验证。
综上所述,基于TCGA数据库,本研究利用生物信息学方法鉴定出UM转移相关的多个lncRNA,并以此构建了ceRNA 调控网络,其中6 个中枢lncRNA与UM总体生存率显著相关,将来仍需要实验揭示它们在UM转移中的具体作用机制。
开放获取声明
本文适用于知识共享许可协议(Creative Commons),允许第三方用户按照署名(BY)-非商业性使用(NC)-禁止演绎(ND)(CCBY-NC-ND)的方式共享,即允许第三方对本刊发表的文章进行复制、发行、展览、表演、放映、广播或通过信息网络向公众传播,但在这些过程中必须保留作者署名、仅限于非商业性目的、不得进行演绎创作。详情请访问:https://creativecommons.org/licenses/by-nc-nd/4.0/。
