甲硫醇催化分解的典型活性中心研究进展
2022-03-03曹小华罗永明赵雨桐张文君陆继长
曹小华,罗永明,赵雨桐,田 睿,张文君,陆继长
(1.昆明理工大学 环境科学与工程学院,云南 昆明 650500;2.昆明理工大学 挥发性有机物污染防治与资源化省创新团队,云南 昆明 650500;3.云南省高校恶臭挥发性有机物控制重点实验室,云南 昆明 650500)
0 引 言
甲硫醇(Methyl mercaptan)作为含硫挥发性有机物(VOSCs)的典型代表之一,具有嗅觉阈值低[1](体积分数0.4×10-9)、易挥发、高毒和高腐蚀的性质[2-4],在常温下是一种无色、有烂洋葱味的气体。吸入少量甲硫醇会刺激人类的中枢神经系统;高浓度会引起呼吸麻痹,严重致死[5]。排放到大气中的甲硫醇还会在阳光作用下发生一系列化学反应,导致光化学烟雾、酸雨、臭氧层破坏等环境问题的产生[6]。另外,甲硫醇的来源非常广泛,主要来源包括能源相关的事件(如天然气和石油生产过程)、蛋氨酸的合成以及废水/污水处理和卫生填埋等[7-9]。因此,开发一种有效的甲硫醇去除技术对环境保护具有重要意义。目前报道的脱除甲硫醇的方法有催化分解法[10-17]、催化氧化法[18-23]、碱液吸收法[24-25]、生物处理法[26-27]等。其中,催化分解法由于其操作简单、高效、快速、不需要额外的添加剂而且产物多为小分子物质[28-29]等特点而受到广泛关注[10-11,24]。但是,目前应用于甲硫醇催化分解的催化剂均存在碳中毒或者硫中毒的问题[16-17,30]。已报道文献对催化分解甲硫醇的反应机理和失活机理尚未研究透彻,对活性中心的认识尚且不够。据文献报道,现有催化分解甲硫醇的活性中心主要有活性氧物种、氧化还原位点和酸碱位点三类活性中心。本文主要结合前人研究,总结了目前在催化分解甲硫醇体系中存在的几种活性中心以及对应的反应机理和失活机理,同时介绍了通过添加不同金属助剂和替换载体的方式来调控催化剂的活性中心,以期为后续抗硫、抗碳催化剂的设计提供可借鉴的思路。
1 催化分解甲硫醇的研究现状及进展
1.1 甲硫醇的处理方法
国内外已报道的去除甲硫醇的方法主要归类为吸附/吸收法、生物法和催化降解法。
吸附法包括物理吸附法和化学吸附法。物理吸附法[31]主要利用具有巨大比表面积的活性炭等吸附剂将甲硫醇吸附在孔道内实现去除目的,但是这种方法不适用于高浓度甲硫醇的去除。化学吸附法[32]通常利用吸附剂(活性炭和沸石分子筛)表面的活性基团如羟基、羧基等与甲硫醇分子产生化学作用,生成化学键引起吸附,从而达到去除甲硫醇的目的。其中,沸石分子筛[33-35]利用其优异的孔道结构和晶型结构吸附甲硫醇。然而,化学吸附法同样不适用于高浓度的甲硫醇气体,而且吸附剂性能严重影响吸附效果。吸收法常见的应用于甲硫醇去除的是溶液吸收法[36-37]。该法是将酸性甲硫醇气体通入到溶液中,通过溶液中碱性物质吸收或者氧化剂氧化去除。但是此法产生的吸收液会造成二次污染,增加了再处理成本。
生物法[38]是指通过微生物的降解作用将甲硫醇转化为二氧化碳和水等简单的无机物,达到去除甲硫醇的目的。然而生物法中所需的菌种培养条件较为严格,易受pH、湿度和温度的影响。此外,微生物降解甲硫醇周期长,经济成本高,占地面积大,因此该法不适合推广到工业应用。
催化降解法包括催化氧化法、光催化氧化法和催化分解法。催化氧化法[39-40]是利用氧化剂(氧气、空气以及过氧化氢等)与甲硫醇反应,然后催化氧化生成硫酸盐和甲基硫醇盐等物质。光催化氧化法[41]是通过太阳光激发催化剂而引起氧化-还原反应去除甲硫醇。光催化氧化法去除甲硫醇的主要产物为亚硫酸酯、硫酸酯、亚磺酸酯等含硫物质。催化氧化和光催化氧化的含硫产物都会容易造成二次污染,需要进一步处理。其中,催化分解法[24]将甲硫醇直接分解为简单的小分子产物,如甲烷和硫化氢等。其中,硫化氢可以采用成熟的克劳斯工艺去除。因催化分解法操作简单、不需要额外的添加剂、产生的污染物较少等优点而得到了广泛的研究。
1.2 典型的固体催化剂体系
1.2.1 固体碱催化剂
固体碱催化剂的活性中心是一个表面阴离子空穴,由表面O2-、O2-和—OH组成它的自由电子中心,具有极强的供电子或接受电子的能力[42]。处理甲硫醇的固体碱催化剂包括高分子/无机固体碱。AL German[43]和Schut ten J[44]分别用胺基(—NH2)基团修饰过的聚乙烯酰胺和聚丙烯酰胺络合磺化酞菁钴合成高分子固体碱催化剂用于硫醇的催化分解。催化剂上碱性的—NH2基团可以吸附酸性的硫醇分子,且—NH2基团数量和反应活性成正相关。此类催化剂缺点是高分子载体的溶胀性会致使活性组分团聚,进而使催化活性降低,且催化剂存在再生难的问题。无机固体碱催化剂主要分为金属氢氧化物、金属氧化物固溶体和负载型固体碱[45]三类。以金属氧化物固溶体为例,美国UOP公司[46]研究发现复合金属氧化物固体碱催化剂(MgO/Al2O3、NiO/MgO/Al2O3和NiO/Al2O3)主要利用其表面的碱性中心为脱除甲硫醇的过程提供碱性环境,促使催化反应正常进行。但是,对于酸性硫醇分子的吸附能力较弱,且物理吸附为主,化学吸附较少。此外,梅华[47]等人发现MgO/Al2O3-CoPcS催化剂具有更多的碱性中心有利于硫醇的分解,但是其产物包括二甲基硫醚和二甲基二硫醚等新的有机含硫污染物,存在二次污染的问题。
1.2.2 固体酸催化剂
固体酸催化剂[48]是通过存在于催化剂固体表面上的具有催化活性的酸性中心断裂甲硫醇的C—S键来去除甲硫醇。沸石分子筛催化剂为目前应用于催化分解甲硫醇的主要固体酸催化剂,其可以在非临氢条件下使甲硫醇转化为甲烷、硫化氢等简单的小分子产物。甲烷可以用作燃料或者通过低温甲醇洗实现与硫化氢的分离,分离后得到的甲烷可用作甲烷干重整的反应气,实现碳减排。硫化氢可以采用较为成熟的克劳斯及改进克劳斯工艺进行硫回收。Vasile Hulea[24]等人研究了H-ZSM-5、H-Y、H-FER、H-BEA、H-MOR和H-SAPO-34沸石分子筛催化分解甲硫醇的过程。研究发现,具有拓扑结构的H-ZSM-5分子筛表现出最佳的催化活性和稳定性。在823 K温度条件下,H-ZSM-5催化分解甲硫醇的转化率可达到100%,产物主要为甲烷和硫化氢等小分子产物。然而,该沸石分子筛酸性强,催化剂易积碳,稳定性极差(不足6小时)。
2 现有的催化分解甲硫醇活性中心
2.1 活性氧物种
活性氧主要通过氧硫替换原理,即利用晶格氧、氧空位等活性氧物种中的氧元素替换甲硫醇分子中的硫元素,实现甲硫醇的催化分解。氧化铈[49-54]由于具有丰富的氧空位可为诸多反应提供氧源,而被广泛应用于多个体系。N Laosiripojana[11]等人采用阳离子表面活性剂辅助法合成超细纳米氧化铈,至少需要20个小时才可成功制得所需催化剂。通过OSC表征证明所合成氧化铈具有极强的氧化还原性,又通过H2-TPR和18O/16O同位素交换方法测得氧化铈上晶格氧具有较高的迁移率。此氧化铈中含有高浓度的高流动性氧空位,这些空位是其表面发生反应时氧的提供源。在反应过程中,氧化铈可将甲硫醇转化为小分子甲烷和硫化氢等产物。但是不足之处在于,此方法合成的纳米氧化铈需在高温900 ℃才可实现甲硫醇的完全分解,转化温度较高,能耗较大,不适用于工业应用。
He[16,55]等采用微波辅助络合法合成纳米氧化铈,此法仅需2个小时即可快速合成。与上述阳离子表面活性剂辅助法相比,大大缩短了材料合成时间。该方法可形成更多的晶格氧物种和结构缺陷,有利于甲硫醇的高效催化分解。通过活性测试发现,在450 ℃即可实现甲硫醇的完全分解,且转化温度大大降低。具体反应过程如图1所示,酸性分子甲硫醇首先吸附在氧化铈催化剂的碱性位点上,然后由催化剂上的活性氧(晶格氧、氧空位等)通过氧硫替换原理分解甲硫醇,将其转化为CH4、H2S产物。通过XRD和XPS表征反应一定时间段后的和完全失活的催化剂,发现催化剂表面生成大量的Ce-O-S和Ce2S3物种。主要原因归为氧化铈催化剂表面的活性氧中的O与甲硫醇分子中的S发生了替换,从而生成了Ce-O-S和Ce2S3物种,覆盖在催化剂表面并消耗大量的活性氧,最终导致催化剂失活。通入空气再生可将Ce3+氧化为Ce4+,进而使其恢复到新鲜催化剂的状态,可实现多次循环再生。但是由于催化剂的Ce2S3累积和晶格氧消耗导致催化剂稳定性较差(不足10个小时),工业化过程长期运行需多次再生,工艺较为复杂繁琐。

图1 氧化铈催化剂催化分解甲硫醇机理图[16]
克服上述纳米氧化铈催化剂稳定性差的一种方法是在其表面掺杂适量的金属离子,特别是一些具有不同离子半径的稀土阳离子[56-57]。一方面,当引入低价态金属离子时,掺杂阳离子随机取代部分Ce4+离子。此时,遵循电荷补偿机制,由于掺杂的稀土金属阳离子与Ce4+半径不同,氧化铈基催化剂会产生晶格畸变,形成氧空位[58-59]。另一方面,掺杂离子半径较小的稀土金属阳离子到氧化铈晶格中,同样可以形成更多的表面缺陷及氧空位,进而提高催化剂的储氧能力(OSC)和催化性能[60-61]。基于此,何[10,62]等人通过在氧化铈基材料中(Ce4+离子半径为0.97 Å)引入三价稀土金属阳离子如Y、Gd、Sm、Nd和La等(离子半径依次为1.03、1.05、1.08、1.11、1.15 Å)[63-64],改善催化分解甲硫醇的催化性能。掺杂的稀土金属离子额外提供了大量的碱性位点[65]、氧空位和晶格氧,有利于进一步提高稀土复合氧化铈基催化剂催化分解甲硫醇的活性。陈[30]等人采用微波辅助柠檬酸络合法在CeO2中掺杂一定量的ZrO2,可形成特定结构及颗粒度的铈锆固溶体复合氧化物(CexZr1-xO2),不仅有效地阻止CeO2烧结,而且能够提高铈锆固溶体的储氧能力、低温催化活性和热稳定性。然而,何和陈的改性工作并没有明显改善氧化铈基催化剂的稳定性(<20小时)。究其原因,主要是因为氧化铈基催化剂表面的高浓度活性氧在催化分解甲硫醇的过程中,不仅会捕集甲硫醇分子中的S,还会捕集产物硫化氢中的S,使得催化剂表面生成大量的Ce2S3物种,导致催化剂硫中毒而失活。
2.2 氧化还原位点
理论上来讲,变价金属一般都具有一定的氧化还原性,而氧化还原性一般伴随着晶格氧的变化,进一步影响催化剂的性能。目前报道的以氧化还原位点作为催化分解甲硫醇的活性中心的催化剂,主要有铬基[12,13,66-68]和铈基[10,15-16,55,62]催化剂。铬/铈物种的存在能够通过Cr(Ⅵ)/Ce(Ⅳ)和Cr(Ⅲ)/Ce(Ⅲ)之间的氧化还原循环增强催化剂的还原性。以铬基催化剂为例,陆[67]等人采用等体积浸渍法制备的不同负载量的Cr(x)-Al2O3(质量比x=1.0%,2.5%,5.0%,7.5%和10%)催化剂展现出良好的催化性能,显著降低了活化温度。在低温375 ℃,Cr(7.5)-Al2O3催化剂分解甲硫醇的转化率就可以达到100%。通过XPS表征测试反应前后的Cr(7.5)-Al2O3催化剂,新鲜的催化剂中Cr6+和Cr3+的含量分别为63.2%和36.8%(如图2所示)。稳定性试验25小时后,Cr(7.5)-Al2O3中Cr6+种类的百分比下降到23.6%,而Cr3+占76.4%。稳定性测试40小时后,XPS只检测到Cr3+种,表明Cr(7.5)-Al2O3表面的Cr6+种完全转化为Cr3+种。这一结果也表明了除Cr3+外,Cr6+物种在CH3SH分解过程中起关键作用,即证明Cr6+是催化分解甲硫醇的活性物种。

图2 Cr(7.5)-Al2O3催化剂的Cr 2p XPS谱图[67]
2.3 酸碱位点
碱性中心用于吸附酸性的甲硫醇分子,酸性中心比如强B酸或者具有L酸酸性的金属离子,均可通过断裂C—S键实现甲硫醇的分解。据Edouard Huguet[24]等人报道,沸石分子筛如H-ZSM-5、H-Y和H-镁碱沸石等可将有机硫物种如甲硫醇分子催化分解为碳氢化合物和硫化氢小分子产物,无需额外添加剂,如图3所示。其中硫化氢可采取成熟的克劳斯及改进克劳斯工艺实现硫回收。H-ZSM-5催化分解甲硫醇的在线反应寿命非常短,不足6小时便开始快速失活。通过TGA表征分析,发现在反应6小时后的催化剂上就明显检测到了大量积碳,因此推断失活原因是由于催化剂表面累积大量积碳覆盖了强酸性中心。而H-ZSM-5催化剂的强酸性中心是影响催化剂活性和稳定性的重要因素,甲硫醇首先吸附在催化剂的碱性中心上然后再由强酸性中心断裂C—S键达到分解目的。但是强酸性过强就会使得C—S键更容易断裂,从而导致更多的积碳累积在催化剂表面或者堵塞催化剂微孔孔道,造成其催化性能下降。
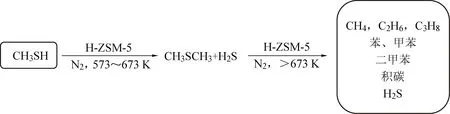
图3 H-ZSM-5催化剂催化分解甲硫醇机理图[24]
3 不同金属助剂/载体对催化分解甲硫醇活性中心的调控
3.1 不同金属助剂对活性中心的调控
据前人文献报道可总结出,调控晶格氧浓度、增强催化剂的氧化还原性以及适量的增加碱性中心的数量、减少强酸性中心的数量都有助于催化分解甲硫醇性能的提高。基于此,部分学者制备了系列改性后的催化剂用于恶臭甲硫醇气体的催化分解研究。
陈[15]等人设计了一系列的H2-TPR实验用于证实晶格氧对催化分解甲硫醇起主要作用而非体相氧物种。因此,何等人通过向氧化铈中掺杂不同离子半径的稀土金属(Gd、Sm和Nd)[62]和三价稀土金属(Y、Sm和La)[10]制备成Ce0.75RE0.25O2-δ金属复合氧化物,改性后的Ce0.75Gd0.25O2-δ催化剂增加了适量的活性中心如碱性中心、晶格氧和氧空位,有利于吸附更多的甲硫醇分子并对其进一步催化分解。而Ce0.75Nd0.25O2-δ催化剂由于碱性中心过多,在反应中它不仅吸附甲硫醇分子而且会吸附大量的硫化氢分子,占据更多的活性中心。随着反应的进行,催化剂上累积了大量的硫化铈物种(Ce2S3),其会消耗和覆盖大量活性中心,最终导致催化剂失活。后期,陈[15]等人采用微波辅助柠檬酸络合法制备的Ce1-xYxO2-δ固溶体催化剂具有更佳的催化性能(如图4所示)。一方面,主要由于Y的添加,使得晶粒尺寸减小,缩短了晶格氧迁移的距离,有利于体相晶格氧向表面的快速迁移。另一方面,Y进入氧化铈的晶格,形成更多的结构缺陷,提高了氧迁移率。以上两方面都有利于催化剂表面消耗的活性中心晶格氧得到快速补充,因而使催化剂稳定性有所改善。但是最佳的铈基催化剂的稳定性目前也没有超过20小时。总体来说,对于氧化铈基催化剂容易硫中毒而导致稳定性差的原因是铈基催化剂颗粒尺寸大、高温易团聚和活性氧浓度过高,易捕集过多的含硫物种。因此,后期改性铈基催化剂的方向应该是减小氧化铈颗粒尺寸和高分散活性相金属铈,从而减缓催化剂硫中毒进而提高催化剂稳定性。

图4 Ce1-xYxO2-δ和CeO2催化剂催化分解甲硫醇机理图以及催化性能图[15]
强酸性位点是催化分解甲硫醇的活性中心之一,但是强酸性过强则会断裂更多的C—S键,导致催化剂深度积碳而使催化剂失活。因此,减少强酸中心数量是解决固体酸催化剂碳中毒的重要策略之一。已有报道,部分学者通过添加稀土金属和碱金属等助剂来调控催化剂的酸碱性中心数量。陆[67]等人制备了稀土金属La改性的H-ZSM-5催化剂应用于甲硫醇的催化分解。其于450 ℃即可完全分解甲硫醇,在线反应90小时后甲硫醇转化率仍保持在95%,稳定性是H-ZSM-5的9倍。通过XPS、XRD和FT-IR表征证明,13% La/HZSM-5催化剂表面生成的大量氧化镧、碳酸氧镧等碱性镧物种不仅可以提供碱性中心吸附酸性甲硫醇分子,而且可消除和抑制积碳,进而减弱催化剂碳中毒的程度。同时,改性后的La/HZSM-5催化剂的强酸中心数量显著减少,抑制了催化剂表面和孔道内积碳的形成。余[69]等人采用等体积浸渍法制备了碱金属Na改性的H-ZSM-5催化剂,并对其催化去除甲基硫醇的活性进行了分析。结果表明,适量Na的引入提高了H-ZSM-5的催化活性,显著降低了CH3SH完全转化的反应温度,3%负载量的Na改性H-ZSM-5的催化活性远高于未改性的H-ZSM-5,主要原因是碱金属Na的引入增加了催化剂的碱性中心,利于酸性甲硫醇的吸附。但是Na的负载量过高,将会破坏H-ZSM-5催化剂的骨架结构,而导致催化剂性能变差。
3.2 不同载体对活性中心的调控
此外,随着对高性能催化剂需求的不断增加,研究人员发现载体不仅是活性金属的支撑体,同时载体的酸碱性、氧化还原性及其与金属之间的相互作用等均可显著影响催化剂的反应性能。赵[12]等人发现许多载体通过改变表面CrOx结构中Cr-O键与载体之间的界面性质,以此影响铬物种在催化分解甲硫醇反应体系中的催化性能。在众多载体中,Al2O3和SiO2由于具有惰性、低成本和大比表面积的优良特征,被认为是不错的备选载体。赵[12]等人采用等体积浸渍法制备了xCr/AlO和xCr/SiO催化剂用于甲硫醇的催化分解性能研究。xCr/AlO和xCr/SiO催化剂的表面性质和催化性能的结构模型(如图5所示)。用多种技术手段对xCr/AlO和xCr/SiO催化剂进行表征,结果表明,金属-载体相互作用很大程度上取决于载体表面羟基的性质和数量。通过XRD、UV-vis、XPS和Raman表征结果发现,在xCr/AlO催化剂上主要是单聚体的Cr(VI)物种,而在xCr/SiO催化剂上则以团聚的Cr2O3物种为主。H2-TPR和NH3/CO2-TPD结果表明,xCr/AlO相比xCr/SiO催化剂具有更强的氧化还原性和酸碱性。其中,由Cr6+和Cr3+价态变化产生的氧化还原性有利于提高催化剂的低温活性,碱性中心有利于酸性甲硫醇分子的吸附,酸性中心有利于C—S键的断裂。更重要的是,金属-载体的相互作用对掺杂铬的分布、氧化态、还原性和酸碱性起着至关重要的作用。从CO2-TPD、FT-IR和TG表征结果发现,Al2O3载体表面覆盖了大量的羟基,大大增强了金属-载体的相互作用力。界面上的铬和Al2O3载体具有更强的相互作用,有利于活性物种铬的分散性的提高。进而使得xCr/AlO表现出比xCr/SiO催化剂更佳的催化分解CH3SH的性能。可见,载体性质对催化剂活性中心的调控起着至关重要的作用。
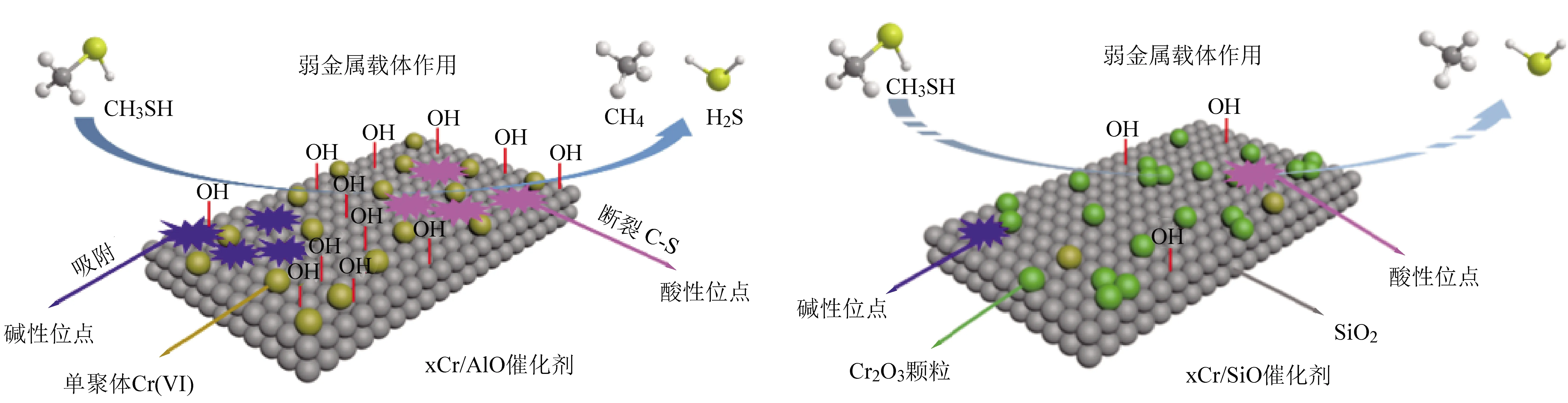
图5 载体表面性质机理及xCr/AlO和xCr/SiO催化剂的催化性能[12]
4 催化分解甲硫醇的反应机理及失活机理
现用于催化分解甲硫醇的催化剂主要利用催化剂的活性氧物种、氧化还原位点和酸碱位点达到分解目的。不同的活性中心对应的催化分解甲硫醇的反应机理不同,而反应机理不同又会导致催化剂表现出不同的失活机理。何[11,15-16,55]等人报道活性氧物种可通过氧硫替换原理实现甲硫醇的去除。以氧化铈基催化剂为例,酸性的甲硫醇分子首先吸附在氧化铈催化剂的碱性位点上,然后吸附的甲硫醇分子迁移到活性中心(活性氧物种)上进行氧硫替换过程。但是活性氧浓度过高,将会使其在捕集甲硫醇分子中的硫的同时还会捕集硫化氢中的硫[10],使催化剂表面累积大量的Ce2S3物种。过多的Ce2S3物种将会覆盖催化剂表面的活性中心,进而使催化剂硫中毒而失活。而氧化还原位点作为催化分解甲硫醇的活性中心主要影响催化剂的低温活性。无论铈基催化剂[10,15-16,55]还是铬基催化剂[12-13,67],Ce4+/Cr6+和Ce3+/Cr3+在进行氧化还原反应的过程中会促进结构缺陷/氧空位的形成。而形成的大量结构缺陷/氧空位则会大大降低甲硫醇和中间体甲硫醚物种的活化温度,进而使得甲硫醇在低温350~400 ℃之间即可实现甲硫醇的完全分解。活性中心酸碱位点催化分解甲硫醇的主要反应机理是:催化剂的碱性中心优先吸附酸性的甲硫醇分子,然后由强酸性中心断裂甲硫醇分子的C—S键,最终将甲硫醇转化为小分子产物得以分解。低于400 ℃,甲硫醇分子在催化剂的作用下断裂S—H、C—S键转化成中间体甲硫醚(CH3SCH3,DMS)和硫化氢(H2S)分子;当温度大于400 ℃时,甲硫醚进一步分解为甲烷(CH4)和硫化氢(H2S)等小分子产物。以H-ZSM-5[24]催化剂为例,强酸性中心虽然是甲硫醇催化分解反应的活性中心,但是强酸性过强将会导致更多的C—S键断裂,而使得大量的碳、硫累积在催化剂表面覆盖活性中心或者堵塞催化剂孔道,造成催化剂碳中毒或硫中毒。以上是目前已经报道的三大活性中心的相应反应机理和失活机理,从中可知三类活性中心都面临碳中毒或者硫中毒的难题。
5 结语与展望
综上所述,从已报道的文献中发现在一种催化剂上会同时存在两种或三种活性中心,因此,三类活性中心在催化分解甲硫醇的过程中属于协同作用。在催化分解甲硫醇的反应中,部分催化剂体系是由于碳中毒或者硫中毒而导致催化剂失活,但是大多数情况是碳中毒和硫中毒同时发生。通常情况下,催化剂碳中毒可通过简单的空气再生实现催化剂初始高活性的再现,而硫中毒再生则会生成大量硫酸盐,而造成催化剂的难再生。因此,对于抗中毒催化剂的设计,提供以下思路:先将催化剂硫中毒问题转化为碳中毒问题,其次通过调控催化剂活性中心等手段,减缓催化剂碳中毒问题。
本文通过对催化分解法去除甲硫醇的研究现状、反应机理及失活机理进行总结,发现可以通过调控活性中心(活性氧、氧化还原位点和酸碱位点)来将催化分解甲硫醇过程中的硫中毒问题转化为碳中毒问题,紧接着后续减缓碳中毒问题,具体方法如下:(1)针对活性氧浓度过高而导致催化剂快速硫中毒的问题,可适当降低活性氧的浓度,比如将金属氧化物分散在高比表面积的分子筛上,可提高活性氧的分散度,以此减少对硫的大量捕集;(2)据文献报道小尺寸的金属颗粒可以抗硫,因此可以通过原位流动气氛处理等方法制备小尺寸的催化剂;(3)适当的添加稀土金属,提供更多的碱性位点,一方面利于酸性甲硫醇分子的吸附,另一方面生成的碱性物种可和积碳反应,达到去除积碳的目的;(4)选择氧化还原性强的活性金属作为活性组分,氧化还原过程中形成的大量氧空位等活性氧物种同样可以减少积碳;(5)适当掺杂稀土金属或者碱金属,调控酸碱位点活性中心数量,避免强酸性过强导致催化剂积碳。以上方法可有效减缓催化分解含硫挥发性有机物体系中催化剂中毒过程且可为抗硫抗碳催化剂的设计提供新思路。
