无机钙钛矿材料力学及电子输运性质调控研究
2022-02-23阎立夫赵伶玲
阎立夫 赵伶玲
(东南大学能源热转换及其过程测控教育部重点实验室, 南京 210096)
无机钙钛矿热电材料因其可基于塞贝克效应进行温差发电而在工业废热、余热的回收利用等领域具有广阔的应用前景[1-2]. 热电材料的热电转换效率一般用热电优值(ZT)进行表征,其表达式为:ZT=S2σT/k,其中S为塞贝克系数,σ为电导率,T为温度,k为热导率. 从该式可以看出,对具有特定热导率的材料而言,提升电子输运性质参数(热电功率因子PF=S2σ)对热电优值和热电转换效率的提高至关重要. 另外,材料在工业加工过程中会受到拉伸、压缩、剪切等应力作用,因此为实现工业规模化加工生产,调控材料力学性质也具有重要的意义.
近年来,研究者们对无机钙钛矿(CsBX3) 材料的电子输运性质和力学性质开展了广泛研究. Xiao等[3]和Yin等[4]发现钙钛矿材料所具有的较高电子输运性能与其B位金属元素的电子轨道中含有s2单电子相关,因此该研究提出了B位为含s2单电子的铅基 (电子轨道为Pb-6s26p2) 钙钛矿材料,如CsPbI3,经研究得出该种钙钛矿材料的电子输运性能较为优异. 之后,Qian等[5]通过实验方法制备了锡基 (电子轨道为Sn-5s25p2)、锗基 (电子轨道为Ge-4s24p2)钙钛矿合金CsSn0.8Ge0.2I3材料,并在实验测量中得出,上述材料具有良好的热电性能,这进一步证明了含s2单电子的B位金属元素在较大程度上影响了材料的电子输运性质. Koliogiorgos等[6]在探究无机钙钛矿材料B位金属元素种类的基础上,考虑了X位卤族元素的变化,依据第一性原理计算了卤素为I、Br、Cl原子的无机钙钛矿CsBX3(B=Ge, Sn, Pb, Ca, Sr) 材料的电子结构及光学性质,得出钙钛矿材料卤族元素种类对电子结构及光电性质具有较大影响,但是该研究并未开展元素种类对电子输运性质的影响研究. 同时,Lee等[7]依据实验及第一性原理,在详细分析了声子特性的基础上,得出不同B位金属元素及X位卤族元素种类下的钙钛矿材料均具有超低的热导率 (其中,CsPbI3热导率为(0.45±0.05) W/(m·K),CsPbBr3热导率为(0.42±0.04) W/(m·K),CsSnI3热导率为(0.38±0.04) W/(m·K)),表明钙钛矿材料元素种类对热导率影响不大. 由此可见,不同B位金属元素及X位卤族元素的无机钙钛矿材料热电转换效率主要取决于其电子输运性质. 因此,通过改变B位金属元素及X位卤族元素对无机钙钛矿材料性能进行调控的方法目前已受到关注,而不同元素对钙钛矿电子输运性质的调控及影响机理还有待进一步研究.
关于无机钙钛矿材料的力学性质,研究者们也开展了一系列的研究工作. Rakita等[8]计算了无机钙钛矿材料CsPbX3(X=I, Br)的力学性质,发现钙钛矿材料力学性质主要与Pb—X键相关,表明无机钙钛矿材料X位元素种类对力学性质具有较重要影响. Feng[9]探究了有机无机钙钛矿CH3NH3BX3材料在采用不同B位金属元素(Pb、Sn)及X位卤族元素(I、Br)时的力学性质,分析得出B、X元素种类(B—X化学键)可较好调控其力学性质. 然而,关于无机钙钛矿材料CsBX3中B、X元素种类对力学性质的影响研究尚未见报道.
综上所述,目前通过B位及X位对无机钙钛矿CsBX3材料进行性能调控的方法已被应用于有机无机杂化钙钛矿材料力学性能和无机钙钛矿电子结构性质领域. 然而,无机钙钛矿CsBX3材料的元素种类对其力学性质和电子输运性质的调控及影响机理还有待进一步研究.
本文应用第一性原理结合玻尔兹曼输运理论,开展了采用不同B位金属元素及X位卤族元素对无机钙钛矿CsBX3材料力学性质及电子输运性质的影响研究,并进一步分析了其影响机理,可为材料性能调控及其他材料的研究提供理论支撑.
1 计算方法及验证
1.1 计算模型与方法
本文的研究对象为无机钙钛矿CsBX3材料,其中,B位金属元素选用含s2电子的Ge、Sn、Pb元素,以保留其较高的电子输运性质;X位卤族元素则为I、Br、Cl元素. 为简化计算量,本文在探究B位金属元素的变化时,卤族元素选用I元素;而在探究X位卤族元素的变化时,B位金属元素采用Pb元素. 另外,本文中钙钛矿构型选用高温立方体结构,以降低构型对称性对于材料性能产生的影响. 因此,构建了无机钙钛矿CsBX3(B=Ge, Pb, Sn;X=Cl, Br, I) 材料几何模,其构型见图1.
本文首先对上述钙钛矿材料进行结构优化以及电子自洽计算,采用基于第一性原理的密度泛函理论 (DFT) 和GGA-PBE赝势泛函. 计算中的平面波截断能 (ENCUT) 为500 eV,高斯展宽因子0.02.布里渊区采用积分为Gamma K点网格,并选取K点网格密度为9×9×9. 电子自洽计算选取更为密集的K点网格,网格密度为16×16×16. 结构优化应用力的收敛标准为-0.01 eV/nm,电子弛豫为能量收敛标准1×10-5eV.
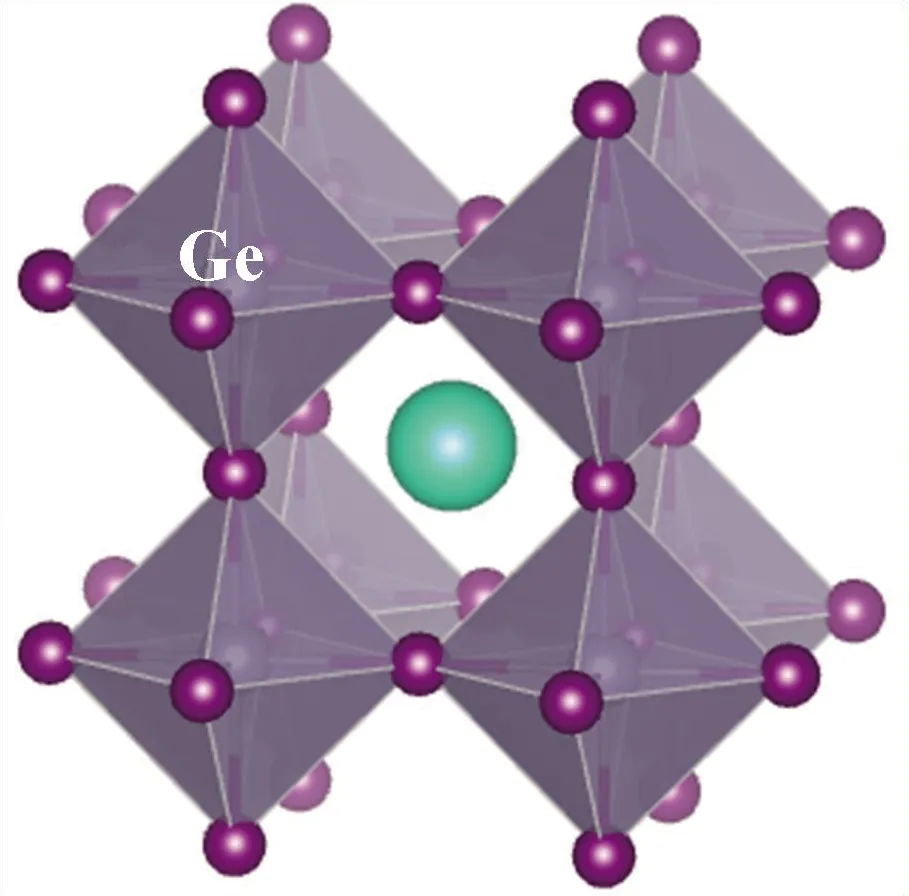
(a) CsGeI3
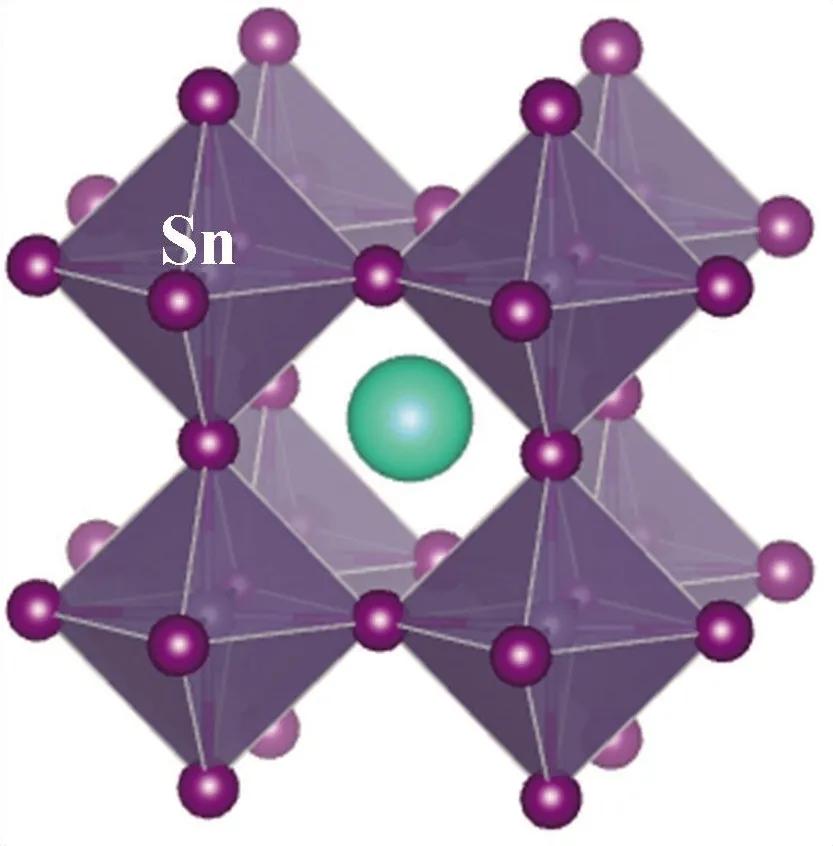
(b) CsSnI3
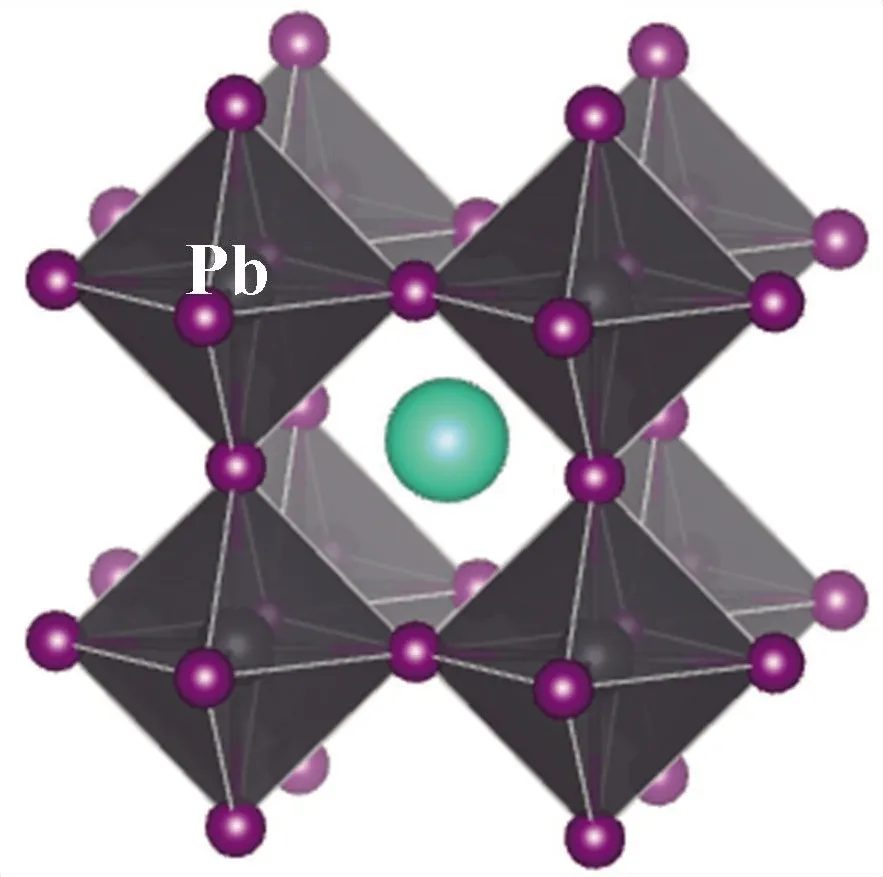
(c) CsPbI3
(d) CsPbBr3
(e) CsPbCl3
针对材料的电子输运性质,本文基于玻尔兹曼输运理论对电子能带进行光滑的傅里叶积分计算. 电子迁移率采用3种模型进行获取,分别是FHIP[12](F)模型、Kadanoff[13](Ka)模型以及Hellwarth-1999[14](H)模型,具体见文献[15]. 本文对电子能带的傅里叶积分是通过BoltzTraP2[16]软件实现的. 应用弛豫时间近似,分别采用形变势理论[17-18]、Brooks-Herring方法[18]及Fröhlich模型[19-21]表征声学声子、杂质离子、光学声子与载流子的散射过程,并针对电子能量进行积分获得平均弛豫时间[22].
1.2 收敛性测试与模型验证
为了验证所建立模型及电子能带结构的准确性和可靠性,计算了CsPbI3钙钛矿材料在K点网格密度为7×7×7、9×9×9、11×11×11下的能带结构,结果见图2. 其中,第一布里渊区K点路径为G-X-M-G-K-X. 从图2中可以看出,K点网格密度在7×7×7、9×9×9与11×11×11时所得能带结构(费米能级均移至0 eV处)基本相同. 此外,所建立的CsPbI3钙钛矿结构晶格常数(0.639 nm)与文献[23-24]报道实验结果(0.629 nm)相近,相对误差在2%以内,因此模拟结果具有可靠性和准确性.

图2 CsPbI3材料电子能带结构收敛性测试
2 结果与讨论
2.1 力学性质
本文首先研究了无机钙钛矿材料的力学性质,包括杨氏模量(E)、体积模量(K)、剪切模量(G)、泊松比(ν) 及德拜温度(T) 等力学性能参数,可为实验中材料的应力调控研究及材料的规模化应用提供理论基础. 本文计算所得的CsBX3(B=Ge, Pb, Sn;X=Cl, Br, I) 钙钛矿材料力学性质参数列于表1. 从表中可以看出,CsBX3无机钙钛矿体积模量范围为17.13(CsSnI3)~27.08(CsPbCl3) GPa;剪切模量范围为8.44(CsPbI3)~13.07(CsPbCl3) GPa;杨氏模量范围为21.74(CsPbI3)~33.71 (CsPbCl3) GPa. 由此可见,上述力学参数的变化范围较大,表明通过卤族元素及金属元素的置换进行力学性质调控具有可行性. 另外,从表1中不难发现,在相似的构型和组成条件下,随着X位卤族元素原子半径的减小(元素原子半径从大到小依次为I、Br、Cl),晶格常数(L)逐渐减小,力学性能参数(E、K、G)逐渐增大. 经分析,这是由于晶格常数越小,元素间杂化程度越高,导致材料弹性常数越大(即被压缩和拉伸越困难),进而造成了材料力学性质的增大. 本文计算了钙钛矿材料的德拜温度(见表1),该数值也可证明力学性质与晶格常数的关联性. 随着晶格常数变大,德拜温度逐渐降低 (其中德拜温度可衡量化合键的强度),表明了共价键杂化程度的减弱. 另一方面,在B位金属元素的变化过程中,钙钛矿的力学参数与B位金属元素原子半径之间也基本呈现相反的变化规律,与X位卤族元素变化情况相似. 但是,CsSnI3的体积模量低于CsPbI3的体积模量,这不符合上述规律,经分析,这主要是由于Sn元素的特殊能级位置及B位金属元素的电负性所造成的. 综上,CsBX3钙钛矿的力学性质主要由B和X元素类型所决定(B2+和X-离子的半径和电负性相关),与Feng[9]探究的CH3NH3BX3杂化钙钛矿结论相似,因此B位及X位元素的置换可较好实现钙钛矿力学性质的调控.
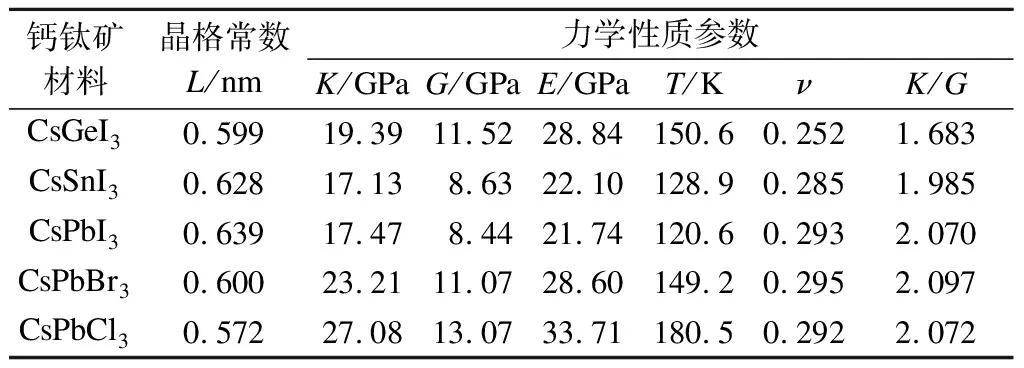
表1 无机钙钛矿材料晶格常数及力学性质参数
为了衡量材料的延展性,本文对材料的体积模量与剪切模量之比(K/G)进行了计算,进而评估材料在加工过程中的受力对材料的影响. 从表1中可以发现,除无机钙钛矿CsGeI3外,CsBX3钙钛矿的K/G数值接近或大于2(一般而言,对于具有良好延展性的材料该数值通常应大于1.75[9]),表明钙钛矿材料具有良好的延展性,即在受压缩、拉伸等外力下能够碾成薄片而不破裂,延伸成细丝而不发生断裂. 由此可见,钙钛矿材料可进一步通过施加应力 (施压、退火、外延晶格失配等手段) 进行材料的性能调控优化. 此外,表1所列钙钛矿材料的泊松比也可证明钙钛矿材料具有良好的延展性 (具有良好延展性固体材料的泊松比通常大于0.26[9]). CsGeI3泊松比为0.252,属于通常的离子或离子共价晶体,其余无机钙钛矿材料的泊松比可达0.285~0.295之间,更类似于分子晶体,且其延展性可与钢材 (泊松比为0.27~0.30)延展性相媲美,进一步证明钙钛矿材料具有可规模化应用的广阔前景.
为衡量材料力学性质的各向异性程度,本文计算了钙钛矿材料三维空间中的力学性质. 三维空间中杨氏模量的分布见图3. 从图3中可以看出,5种钙钛矿材料主轴方向[1 0 0]、[0 1 0]、[0 0 1]的杨氏模量基本相同,经分析这主要是由无机钙钛矿材料的中心对称性所造成的. 同时从图3中可以看到,晶体主轴方向的杨氏模量均大于各自晶体其余方向的杨氏模量,呈现较明显的空间各向异性,表明无机钙钛矿材料更易于承受来自主轴方向的应力作用. 进一步地,5种钙钛矿材料的各向异性程度从大到小依次为CsPbI3、CsSnI3、CsPbBr3、CsPbCl3、CsGeI3,表明X位卤族元素及B位金属元素的置换也可实现力学性能各向异性性质的调控.

(a) CsGeI3

(b) CsSnI3

(c) CsPbI3

(d) CsPbBr3
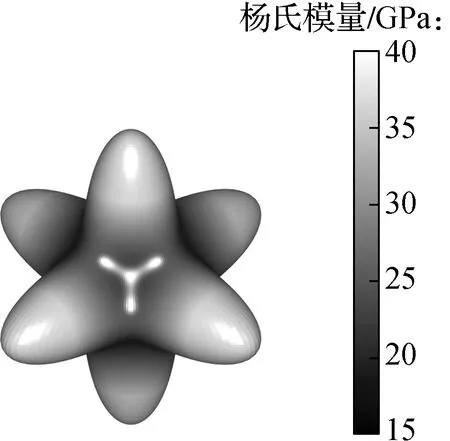
(e) CsPbCl3
2.2 电子结构
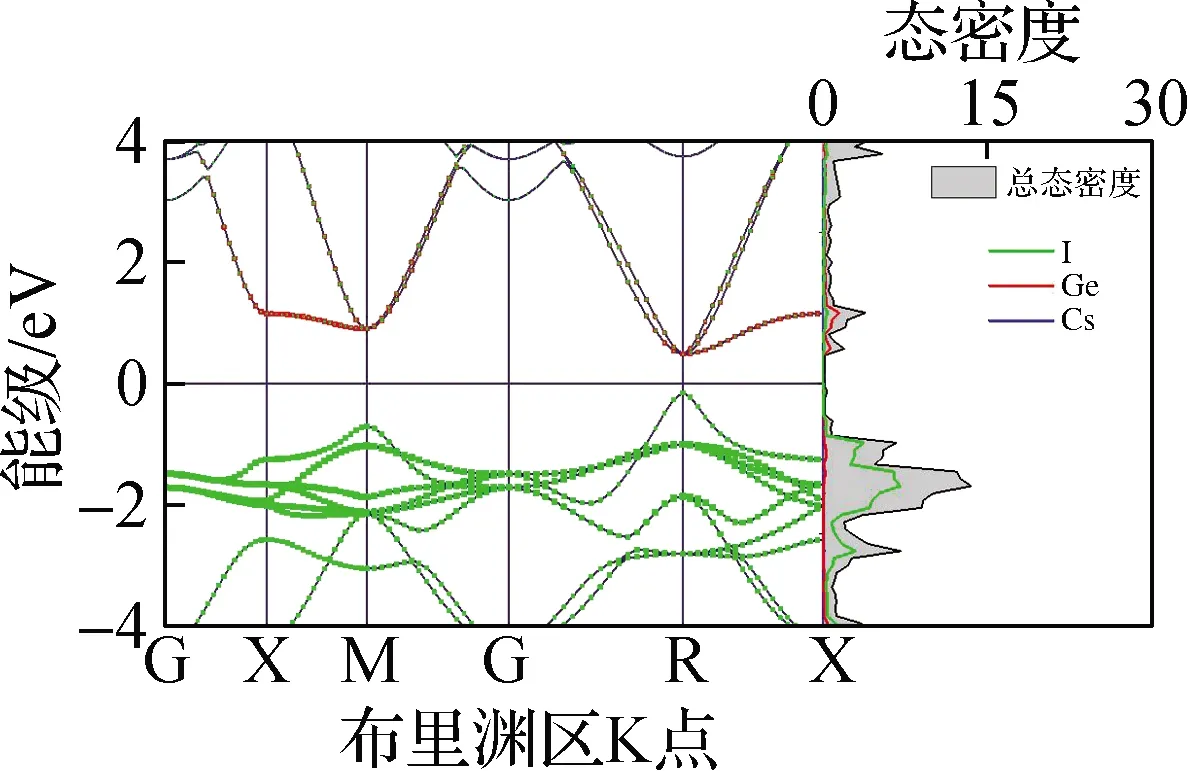
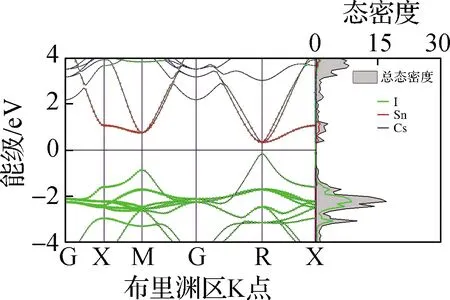
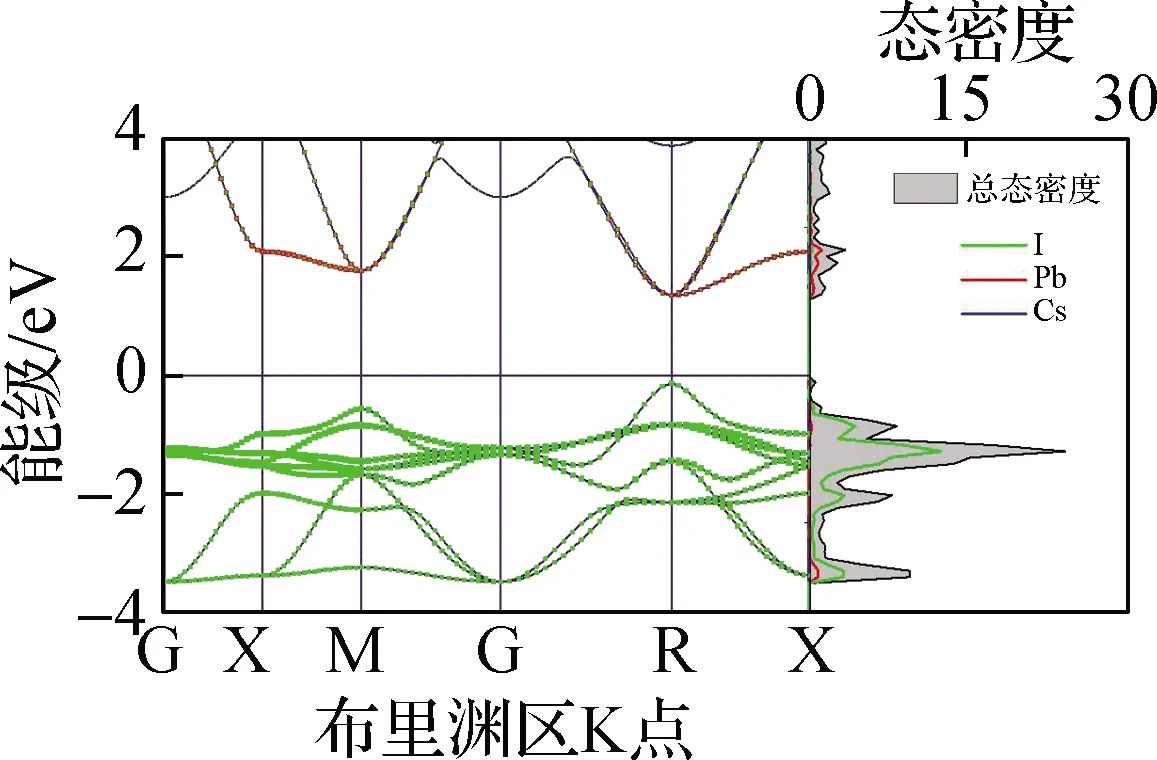
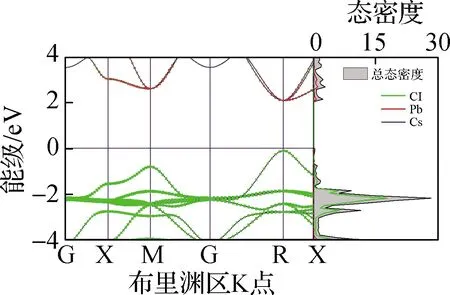
本文基于第一性原理分析了5种钙钛矿材料的电子分布情况,包括电子能带结构、态密度及禁带宽度,结果见图4. 从图4(a)~(e)电子能带结构中可以看出,5种无机钙钛矿材料均为直接禁带宽度半导体,导带底和价带顶均在R点. 进一步地,从态密度中可以看出,导带底(CBM)主要由B-p轨道构成,价带顶(VBM)主要由B-s和卤族X-p轨道杂化构成,且CBM在R点处px、py、pz轨道三重简并,这与文献[25]报道结果相同. 从图4(f)禁带宽度可以看出,CsBX3钙钛矿禁带宽度范围为0.47(CsSnI3)~2.20 eV(CsPbCl3),较大的禁带宽度变化范围表明,通过元素置换对禁带宽度进行调整是一种较为行之有效的调控手段. 另外,随着X位卤族元素原子半径的减小,禁带宽度逐渐增大,经分析得出这主要是卤族元素电负性从I至Cl元素逐渐增强所导致的,即元素越轻电负性越强,导致了禁带宽度越大. 与此同时,键长的减小会导致B-X杂化程度的升高,因此升高了VBM能级,这在一定程度上缓解了由于电负性增强造成的禁带宽度增大的趋势. 同时,在B位金属元素的变化中,CsBI3钙钛矿中B位元素从Pb到Sn,禁带宽度逐渐减小;从Sn到Ge,禁带宽度增大,与文献[26]结果相同. 其中,禁带宽度减小的原因是元素原子半径的减小造成B—X键长的减小,导致杂化程度的升高从而VBM升高. 而禁带宽度增大的趋势主要与B-s能级位置相关,由于Sn-5s与I-5p的能级相比于Pb-6s、Ge-4s与I-5p轨道能级而言更为接近,造成Sn-5s与I-5p杂化程度最高,因此VBM显著上升致使CsSnI3禁带宽度最小. CsSnI3的能级位置及电负性导致其禁带宽度最小,与上文CsSnI3具有特殊的力学性质结论相吻合.
(a) CsGeI3能带结构
(b) CsSnI3能带结构
(c) CsPbI3能带结构

(d) CsPbBr3能带结构
(e) CsPbCl3能带结构

(f) 禁带宽度

(g) 电子平均有效质量
本文基于上述电子结构计算了5种钙钛矿材料的电子平均有效质量(m*),以进一步定量描述其电子结构. 电子有效质量是通过拟合导带底的二阶导数求得. 从图4(g)电子平均有效质量中不难发现,无机钙钛矿的电子平均有效质量均较小(0.13~0.21),表明其具有良好的电子输运性能和较大的电子迁移率. 随着卤族元素原子半径逐渐减小,有效质量逐渐增大;而随着B位金属元素原子半径的减小,电子有效质量先升高后降低. CsSnI3钙钛矿的电子有效质量在B位金属元素变化的3种钙钛矿中最大,导致其呈现了较为异常的输运性质.
2.3 电子输运性质
应用弛豫时间近似计算了5种钙钛矿结构的输运性质随N型/P型载流子浓度(ne/nh)的变化关系,以探究不同B位金属元素及X位卤族元素对无机钙钛矿材料输运性质造成的影响. 由于计算所得N型电子输运性质较P型输运性质更为优异,因此本文主要讨论N型半导体性质. 本文中计算的输运性质主要包含塞贝克系数(S)、电导率(σ)以及热电功率因子(PF=S2σ). 在计算中,通过费米能级的移动得到载流子浓度的变化,以模拟实验过程中的掺杂过程,其中上述输运性质在室温下随载流子浓度的变化关系如图5所示. 从图中可以看出,5种钙钛矿材料的S(绝对值)随着N型载流子浓度(ne)的增大均减小,在较高载流子浓度下,S接近于零;σ随着载流子浓度的增大而增大,与文献[27]结果相同.
针对5种钙钛矿材料的电子输运性质,对于X位卤族元素变化的铅基钙钛矿而言,随着卤族元素原子半径的减小,S在不同载流子浓度下均逐渐增大,但由于σ呈现相反的规律,因此PF随着卤族元素原子半径的减小而逐渐减小,其中CsPbI3钙钛矿PF最为优异. 针对B位金属元素变化的3种钙钛矿材料中,随着B位金属元素原子半径的减小,σ逐渐增大,而S以CsSnI3钙钛矿材料最低,且在较低载流子浓度下S有急剧减小的现象. 这主要是CsSnI3钙钛矿材料禁带宽度最小(小于0.5 eV),在室温下产生了双极化效应所造成的. 综上,B位金属元素变化的3种钙钛矿材料中,以CsGeI3的PF最为优异. 同时不难发现,锡基和铅基钙钛矿材料PF基本相近,这主要是由于锡基钙钛矿较大的电子有效质量造成的,缓解了由于半径变化造成输运性质的升高趋势.
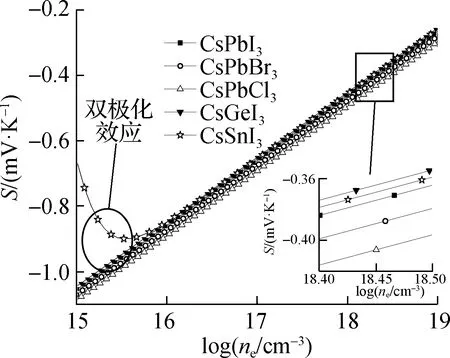
(a) 塞贝克系数
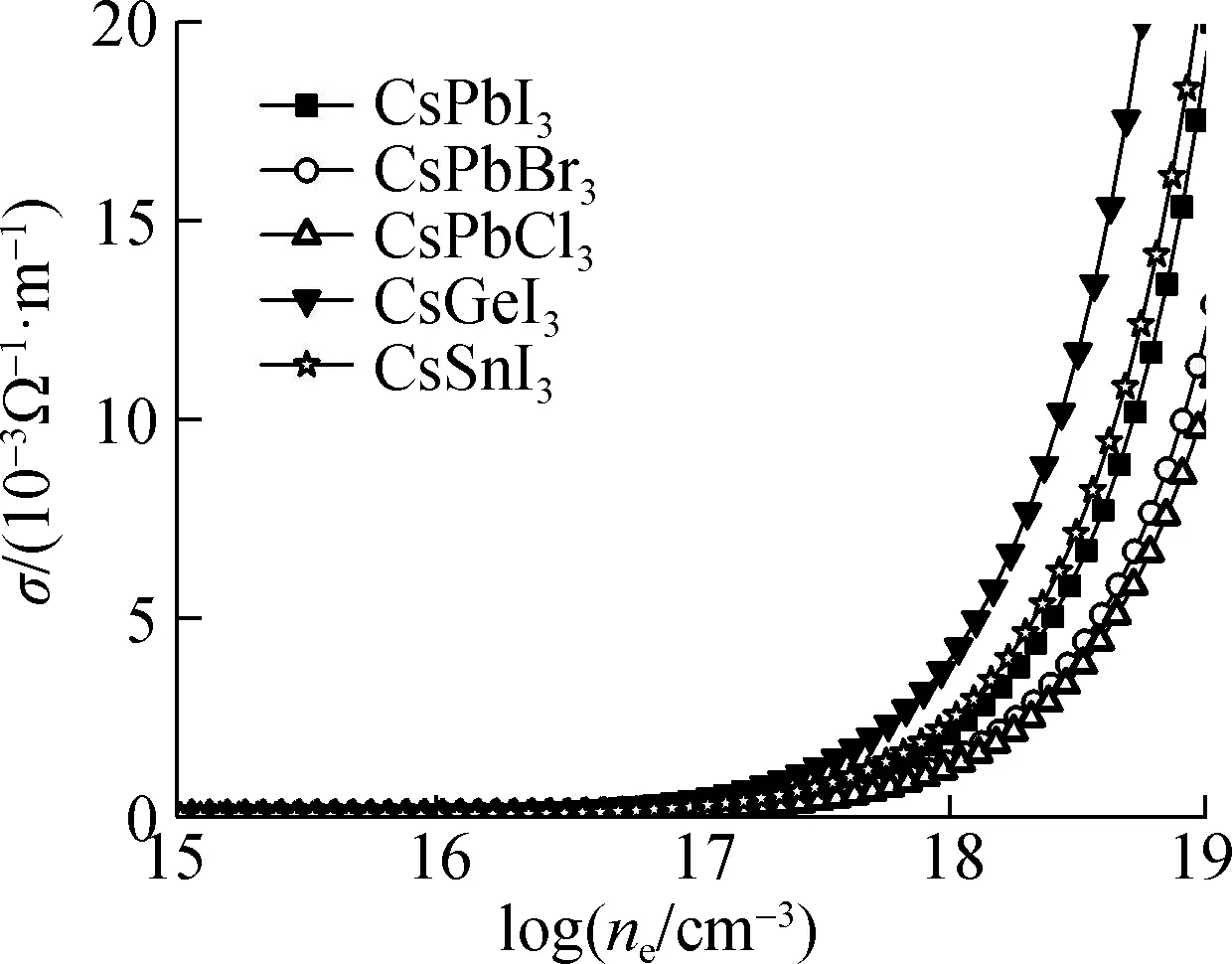
(b) 电导率

(c) 热电功率因子
为进一步描述电子在半导体内部整体的运动快慢程度,本文采用3种模型(F、Ka以及H模型)计算了5种钙钛矿材料的电子迁移率随温度的变化,探究B位及X位元素置换对于电子迁移率的影响. 从图6(a)无机钙钛矿CsPbI3电子迁移率结果中可以看出,对于Ka模型和H模型电子迁移率均随着温度的升高而逐渐减小直至趋于稳定,但对于F模型随着温度的升高,电子迁移率呈现先降低而后升高的趋势,3种模型的变化趋势均与文献[15]结果相同. 其中,应用F模型下计算的电子迁移率随温度的变化趋势,主要是由于F模型采用了低温渐近分析的方法,造成其在高温下与实际数值呈现出较大偏差. 而经文献[15]证明,采用Ka模型和H模型计算的电子迁移率与实验数值吻合较好. 3种模型精度为:H模型精度优于Ka模型精度,优于F模型精度[15],因此本文主要采用H模型数据分析原子种类对电子迁移率的影响. 从图6 (b)X位卤族元素的变化中可以看出,对于H模型,随着卤族元素原子半径的逐渐减小,电子迁移率从CsPbI3至CsPbCl3逐渐减小,这主要是逐渐增大的电子有效质量所造成的. 在室温下由H模型计算可得,CsPbI3电子迁移率最高,可达162 cm2/(V·s),CsPbCl3为42 cm2/(V·s),表明CsPbI3钙钛矿具有良好的电子输运性质,以及X位卤族元素置换可作为调控输运性质电子迁移率的优良手段. 对于图6 (b)B位金属元素的变化情况而言,Sn基钙钛矿与Ge基/Pb基钙钛矿相比呈现出较低的电子迁移率 (120 cm2/(V·s)) ,而CsGeI3电子迁移率最高,对于H模型可达255 cm2/(V·s),表现出良好的电子迁移率. 较大的电子迁移率变化范围(42(CsPbCl3)~255(CsGeI3) cm2/(V·s)),表明通过B位金属元素及X位卤族元素的置换可实现电子迁移率的调控.

(a) CsPbI3钙钛矿电子迁移率(3种模型)

(b) 5种钙钛矿材料电子迁移率(H模型)
3 结论
1) 随着X位卤族元素原子半径的减小,钙钛矿力学性质参数 (体积模量、剪切模量、杨氏模量等) 逐渐升高,电子输运性质参数(PF及电子迁移率等)逐渐降低.在CsPbI3、CsPbBr3、CsPbCl3中,钙钛矿CsPbI3具有最优电子迁移率(室温下可达162 cm2/(V·s)),且具有良好的延展性.
2) 随着B位金属元素原子半径的减小,钙钛矿力学、电子输运性质参数呈现升高趋势,但由于CsSnI3钙钛矿中Sn元素特殊的能级位置,造成CsSnI3钙钛矿材料力学性质及电子输运性质参数低于CsPbI3相应数值. CsSnI3电子迁移率在室温下达120 cm2/(V·s),力学延展性良好.
3) 5种钙钛矿材料中,CsGeI3电子输运性质(PF及电子迁移率)最为优异,在300 K下电子迁移率可达255 cm2/(V·s), 但CsGeI3钙钛矿力学延展性相对较差.
