基于WGCNA鉴定茶树响应草甘膦相关的基因共表达模块
2022-02-22郭永春王鹏杰金珊侯炳豪王淑燕赵峰叶乃兴
郭永春,王鹏杰,金珊,侯炳豪,王淑燕,赵峰,叶乃兴
基于WGCNA鉴定茶树响应草甘膦相关的基因共表达模块
郭永春1,王鹏杰1,金珊1,侯炳豪1,王淑燕1,赵峰2,叶乃兴1
1福建农林大学园艺学院/茶学福建省高校重点实验室,福州 350002;2福建中医药大学药学院,福州 350122
【】分析茶树响应草甘膦相关的基因表达规律和调控途径,在转录水平上探究草甘膦对茶树的作用,确定茶树响应草甘膦的关键基因。以茶树品种‘金观音’为试验材料,将推荐使用浓度的草甘膦施于茶树土壤基质表面,经0、0.25、1、3和7 d后,取叶片进行转录组测序,并测定莽草酸含量。利用WGCNA方法联合分析转录组和莽草酸含量数据,鉴定与草甘膦响应相关的共表达基因模块,筛选关键调控基因。茶树叶片中的莽草酸含量在草甘膦处理后0.25、1和3 d降低,而在7 d时大量积累(为未处理的6.99倍)。从表达谱数据中筛选得到12 568个差异表达基因(DEGs),草甘膦处理不同时间点与未处理数据比对的DEGs均显著富集在苯丙烷、类黄酮生物合成及植物激素信号转导途径;此外,草甘膦处理分别诱导茶树莽草酸代谢及其下游苯丙烷、类黄酮生物合成和激素信号转导途径相关的24、52、31和69个基因差异表达。通过加权基因共表达网络(WGCNA)方法鉴定得到19个基因模块,将转录组与莽草酸含量数据相关联,筛选到两个与草甘膦响应高度相关的关键基因模块,分别包含2 024和2 305个基因。选取关键模块中连通度最高的前50个基因进行共表达分析,获得6个关键调控基因,包括2个抗性基因(和)、1个耐药性基因()、1个离子转运基因()、1个膜转运基因()和1个转录因子()。草甘膦通过干扰茶树叶片中莽草酸代谢,影响其下游代谢途径苯丙烷、类黄酮生物合成及激素信号转导途径的基因转录。此外,研究还鉴定到2个草甘膦响应密切相关的共表达模块,发现、、和等多个潜在候选基因和转录因子在茶树抵御草甘膦逆境中发挥重要作用。
茶树;草甘膦;莽草酸;转录组学;WGCNA
0 引言
【研究意义】茶树[(L.) O. Kuntze]是我国重要的经济作物,其芽叶可制成风味独特的成品茶,具有很大的经济、健康和文化价值[1]。草甘膦是一种最常见的非选择性除草剂,在杀灭杂草的同时,会被茶树根部吸收并转运至全株[2]。草甘膦在茶树体内的残留时间较长,并可能造成茶叶产品中草甘膦的残留量超标,影响茶叶生产安全[3-4]。现阶段,我国福建、江西、安徽的部分茶园已禁用草甘膦,但因其高效廉价,目前在实际生产中完全停止使用草甘膦较为困难。中国农药信息网数据库显示,草甘膦仍是我国茶园主要登记使用的除草剂。因此,深入探究草甘膦对茶树的影响,对保证茶叶质量安全性有重要意义。【前人研究进展】草甘膦主要通过抑制植物莽草酸代谢途径中的关键酶活性,阻止莽草酸向芳香氨基酸(色氨酸、酪氨酸和苯丙氨酸)转化,导致植物体内代谢紊乱[5]。研究证实草甘膦的使用对植物次生代谢物的生物合成存在不利作用,例如,JIANG等[6]研究表明草甘膦除草剂通过抑制大豆莽草酸代谢途径,抑制了大豆顶芽中色氨酸和类黄酮生物合成相关基因的转录;RAINIO等[7]通过对马铃薯的土壤基质进行草甘膦处理,发现植株内的糖苷生物碱浓度下降;MALALGODA等[8]发现使用草甘膦除草剂干扰了小麦体内正常碳代谢,从而对小麦的氨基酸代谢产生负面影响。已有研究表明,草甘膦经茶树根部吸收并富集至叶片,是茶叶中草甘膦残留的主要来源[4,9-11]。此外,笔者课题组前期研究发现,草甘膦的使用不易使茶树出现药害表征,但可显著改变叶片中游离氨基酸、儿茶素和生物碱类化合物的含量[12]。【本研究切入点】目前,草甘膦处理茶树不同时期对叶片基因表达及相关代谢通路的影响有待研究,尤其是基于加权基因共表达网络(weighted gene co-expression network analysis,WGCNA)的茶树响应草甘膦除草剂的基因共表达模块尚未被鉴定。【拟解决的关键问题】本研究对草甘膦处理5个时间梯度(第0、0.25、1、3和7天)的茶树叶片进行转录组测序,分析其基因表达水平和调控途径,并利用WGCNA构建共表达基因模块,与莽草酸数据关联分析,挖掘出与草甘膦诱导相关的关键模块,进而确定模块中与抵御草甘膦逆境相关的核心基因。
1 材料与方法
1.1 材料处理
2019年12月,将一年生‘金观音’茶树(Jin-guanyin)植于体积为2 L的盆钵进行适应性培养,栽培基质为泥炭土(10—30 mm)。于2020年8月选取株高约30 cm、树幅15—20 cm的植株,将5 g·L-1(推荐施用浓度)的草甘膦异丙胺盐在土壤基质表面均匀喷施0.3 L,分别在第0(喷施前)、0.25、1、3和7天采集茶树嫩梢第二叶,液氮速冻,-80℃保存。每个时期取3个生物学重复。
1.2 莽草酸含量测定方法
将参试样品分别在液氮中研磨成粉,称取0.5000 g置于4 mL棕色容量瓶中,加入2 mL去离子水,振荡混匀300 s。60℃水浴超声提取4 h,4 000 r/min离心10 min,取上清液1 mL过0.45 μm滤膜待测。采用高效液相色谱仪(waters E2695)测定样品中的莽草酸含量,色谱条件:C18色谱柱(250 mm×4.6 mm,0.5 μm);柱温:25℃;波长:213 nm;流动相:甲醇﹕1%磷酸水溶液=3﹕97;流速:0.80 mL·min-1;进样量:10 μL。
1.3 转录组测序的获取与分析
采用Plant RNA Purification Reagent(Invitrogen)提取各样本的总RNA,分别使用2100 Bioanalyser(Agilent)和ND-2000(NanoDrop Technologies)检测RNA质量(RIN≥6.5,OD260/280=1.8—2.2,OD260/230≥2.0),琼脂糖凝胶电泳检测RNA完整性。按照Illumina TruSeqTMRNA sample preparation Kit方法构建转录组文库,通过上海美吉生物医药科技有限公司在Illumina Novaseq 6000测序平台上对文库片段进行双末端测序。通过SeqPrep(https://github.com/jstjohn/ SeqPrep)和Sickle(https://github.com/najoshi/sickle)对原始数据进行质控,从而得到高质量的质控数据。利用TopHat 2.1.1软件将质控数据比对到‘黄棪’茶树基因组[13],基因组从中国核酸数据库网站(https:// bigd.big.ac.cn/gsa/index.jsp)下载,GSA:CRA003208。通过Cufflinks软件组装并得到本次与原有注释的差异信息[14],使用RESM软件根据每百万读转录量(TPM)法计算基因表达水平[15]。
1.4 差异表达基因和富集分析
通过EdgeR包(http://www.bioconductor.org/ packages/2.12/bioc/html/edgeR.html)计算筛选差异表达基因(differentially expressed genes,DEGs)[16],差异基因界定为:|log2FoldChange|>1,-value<0.05。差异倍数(FoldChange)表示两样品组间表达量的比值。采用Goatools和KOBAS软件对差异表达基因进行GO和KEGG功能富集分析,当经过校正的值(-value)<0.05时,认为此GO功能和KEGG pathway功能存在显著富集情况[17]。使用Tbtools软件制作热图对基因表达量进行可视化[18]。
1.5 共表达模块构建与分析
利用RESM软件对基因表达数据进行背景校正和标准化,过滤表达量过低和变异系数较小的基因[14]。利用R软件(R version 3.4.4)和WGCNA(R version 1.6.6)包构建基因共表达网络[19],根据过滤数据(平均表达水平1,变异系数0.1)识别与代谢物高度相关的基因模块。过滤后的18 976个基因和3个代谢物的丰度通过计算Pearsons相关性构建共表达网络。使用Cytoscape 3.4.0对核心的共表达模块进行可视化[20]。
1.6 转录组数据验证
采用Primer3plus在线网站(http://www.bioinformatics. nl/cgi-bin/primer3plus/primer3plus.cgi)设计引物(附表1),然后利用全式金Easyscript One-step gDNA Removal and cDNA synthesis superMix试剂盒(北京全式金生物技术有限公司)将RNA合成cDNA。参照王鹏杰等[21]的方法,选用茶树(登录号GE651107)作为内参基因[22],按照Transstart®Tip Green qPCR superMix试剂盒(北京全式金生物技术有限公司)的使用说明在CFX96 Touch荧光定量PCR仪(伯乐生命医学产品(上海)有限公司)上进行qRT-PCR分析,反应程序:94℃ 30 s;94℃ 5 s,60℃ 30 s,40个循环,每个时间点设3次生物学重复。用2-ΔΔCT法计算基因相对表达水平[23],GraphPad Prism 5软件制作柱状图,并通过SPSS 17.0进行差异显著性分析(<0.05)。
2 结果
2.1 草甘膦处理后茶树叶片表型及莽草酸积累量
如图1所示,本试验处理下茶树嫩梢叶片无明显药害特征。图2显示,叶片中莽草酸含量在草甘膦处理0.25 d时显著下降,而后升逐渐升高,草甘膦处理7 d时莽草酸含量最高,达到处理前的6.99倍。

图1 草甘膦处理后茶树叶片的表型

不同小写字母表示差异显著(P<0.05)
2.2 茶树草甘膦处理的转录组分析
2.2.1 测序质量及KEGG富集分析 转录组测序共产生713 187 436个原始序列,在去除测序接头污染和低质量的原始序列后,共获得高质量的706 923 928个过滤序列,共产生104 531 971 833个过滤碱基,过滤序列质量值大于30的碱基所占的百分比(Q30%)在93.69%以上,将各样品的过滤序列与茶树参考基因组比对,比对结果显示各样本的比对率为87.69%—89.79%,序列主要分布在参考基因组外显子区域。转录组测序结果与茶树参考基因组的比对率高,测序质量好,可用于后续分析(表1)。
基于所有样本中的TPM分布情况进行皮尔森相关性分析,结果表明生物学重复样本之间具有很强的相关性,后续分析的结果也更可信(图3-A)。将草甘膦处理不同时间点的数据与未处理的数据比对,共筛选出12 568个DEGs(图3-B)。0.25 d与0 d比对筛选到5 542个DEGs(上调3 319个,下调2 223个);1 d与0 d比对筛选到2 201个DEGs(上调1 308个,下调893个);3 d与0 d比对筛选到2 481个DEGs(上调1 491个,下调990个);7 d与0 d比对筛选到2 344个DEGs(上调1 267个,下调1 077个)。其中0 d vs 0.25 d 、0 d vs 1 d、0 d vs 3 d和0 d vs 7 d比较组中分别有2 971、409、796和879个基因是特有的DEGs;有242个基因在4个比较组中都呈现差异表达,这些DEGs可在草甘膦处理7 d时持续发挥作用。
将4个比较组的DEGs进行生物学代谢途径富集分析,结果仅显示富集程度前10的代谢途径(图4)。与碳水化合物代谢相关的乙醛酸和二羧酸代谢(map00630)仅在“0 d vs 0.25 d”显著富集;与能量代谢相关的硫代谢(map00920)和氮代谢(map00910)仅在“0 d vs 1 d”显著富集;与脂质代谢相关的脂肪酸伸长率(map00062)和-亚麻酸代谢(map00592)仅在“0 d vs 3 d”富集;单萜类生物合成及与氨基酸代谢相关的谷胱甘肽代谢(map00480)仅在“0 d vs 7 d”显著富集。值得关注的是,4个比较组的DEGs均显著富集在苯丙烷和类黄酮等次生代谢物合成(map00940和map00941)及植物激素信号转导(map04075)途径。

表1 参试样品的转录组数据的质量

A:相关性。B:左边水平柱状图表示各集合的DEGs。中间矩阵中,单个点表示某个集合特有的元素,点和点之间的连线表示不同集合特有的交集,竖直柱状图中则分别表示对应交集的DEGs

纵轴表示多个基因集中富集到的KEGG pathway,横轴表示Rich factor(Rich factor越大,表示富集的程度越大),点的大小表示此pathway中的基因个数,点的颜色对应于不同的P值范围
2.2.2 苯丙烷、类黄酮生物合成及莽草酸代谢相关的DEGs分析 茶树在响应草甘膦处理过程中,与苯丙烷、类黄酮生物合成相关的基因在0.25、1、3和7 d均受诱导而差异表达。图5展示了苯丙烷、类黄酮生物合成及莽草酸代谢的通路,以及分别显著富集在各通路上DEGs(24、52和31个)的转录水平。、、、、、和等莽草酸代谢相关的10个基因在处理过程中较高表达(13.85<TPM<179.64)。其中,()、()、()和()在处理各时间点均上调表达,其余6个基因呈现先下降后升高的趋势。、、、、、、、和等苯丙烷生物合成相关的14个基因较高表达(5.28<TPM<353.77),这14个基因在0.25 d下调表达,随后上调表达。、、、、、、和等类黄酮生物合成途径中11个DEGs在处理过程较高表达(4.72<TPM<365.41),并呈现先下调后上调的趋势。
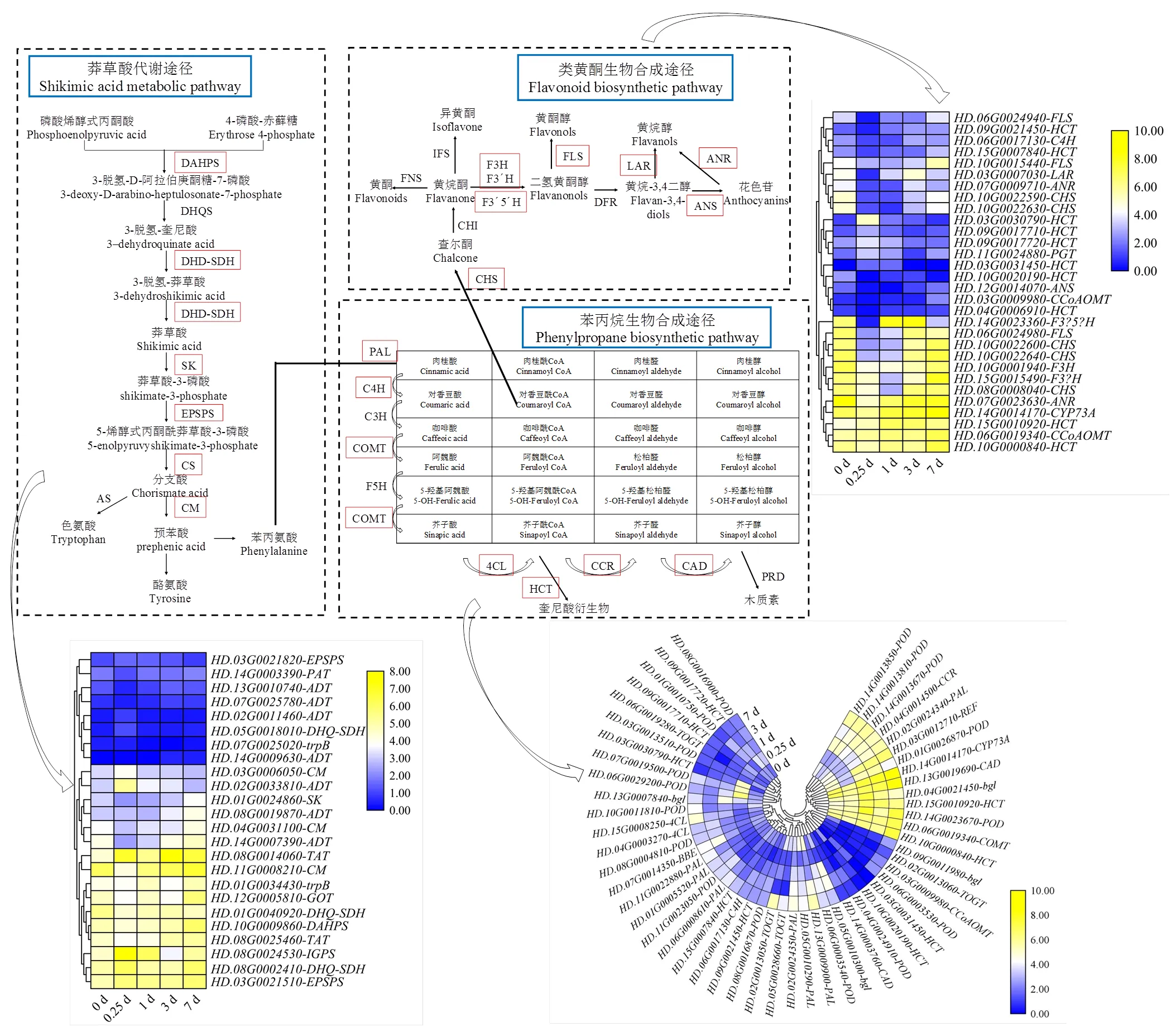
通路图中的红色边框表示各通路上注释到的DEGs;热图表示所有与通路相关DEGs的表达水平,由3个重复样本计算得出的平均值进行log2转换生成。下同
2.2.3 植物激素信号转导途径相关的DEGs分析 草甘膦处理过程中,植物激素信号转导途径中生长素(Auxin)、细胞分裂素(Cytokinine)、脱落酸(Abscisic acid)、赤霉素(Gibberellin)、乙烯(Ethylene)、油菜素内酯(Brassinosteroids)、茉莉酸(Jasmonic acid)和水杨酸(Salicylic acid)等重要防御相关的植物激素信号通路均受到影响(图6-A)。69个基因表达具有差异,并显著富集于植物激素信号转导途径,14个DEGs(,2个;,1个;,1个;,2个;,1个;,1个;,1个;,1个;,1个;,1个;,1个;,1个)在处理过程中较高水平表达(图6-B)。其中,乙烯信号途径下游基因()在处理不同时间点均上调表达(最高上调3.08倍),且表达水平较高(87.88<TPM<271.03);水杨酸信号途径下游基因()在0.25 d显著下调(200倍),而后上调表达(最高上调4.22倍);脱落酸信号途径下游基因()在0.25 d下调表达(5.19倍),而后上调表达(最高上调1.71倍)。
2.3 茶树草甘膦处理的WGCNA分析
2.3.1 基因共表达模块构建 经过滤TPM<1的基因,18 976个差异基因用于构建加权基因共表达网络。通过计算15个样本表达水平的相关系数聚类分析,样本间聚类良好,未出现离群样本(图7-A)。依据无尺度容适曲线处于平滑处,设定幂指数加权值即软阈值为14(图7-B)。
根据差异基因的TPM值进行相关度分析并聚类,相关度较高的基因被分配到同一个模块中,图7-C中聚类树的不同分支代表不同的基因共表达模块,不同颜色代表不同的模块,共划分为19个模块,颜色为灰色代表没有划分到其他模块的基因。
2.3.2 基因共表达模块筛选 图8展示了19个模块各包含的基因个数及其与莽草酸含量的相关性。不同模块间包含的基因个数差异较大,其中,Blue模块的基因个数最多,为3 437个;Royalblue模块的基因个数最少,为60个。相关性系数(绝对值)较大且显著性值较小的模块与表型高度相关,鉴定到与莽草酸极显著正相关的为Green模块(= 0.864,=0.0000329),其次为Brown模块(=0.771,=0.00296);极显著负相关的为Tan模块(=-0.875,=0.0000195),其次为Blue模块(=-0.754,= 0.00117)。
为了解各个共表达模块中基因的生物学功能,对上述各个基因模块进行KEGG富集分析(<0.05),发现Green和Brown模块显著富集到酪氨酸代谢(map00350)、苯丙氨酸代谢(map00360)等芳香氨基酸代谢和花青素的生物合成(map00942)、类黄酮生物合成(map00941)、苯丙烷生物合成(map00940)等次生代谢物合成通路,根据功能关联原则可知这2个基因模块与草甘膦响应相关(图9)。
2.3.3 共表达网络可视化分析 选取Green和Brown模块内连通度最高的前50个基因作为模块的核心基因,并对核心基因的互作网络利用Cytoscape软件进行可视化(图10)。在Green模块中的基因(7 d上调)连通度最高的3个依次是和,是一个植物抗性基因,与614个基因相连;是一种抗病基因,与612个基因相连;是一个是离子转运基因,与608个基因相连。在该网络中发现6个转录因子(,3个;和,1个)与植物防御反应相关。在Brown模块中连通度最高的3个基因依次为、和,是一种多效性耐药性基因,与500个基因相连;是乙烯响应因子,与494个基因相连;是一种膜转运蛋白基因,与480个基因相连。转录因子与植物防御反应相关。
2.4 qRT-PCR验证
为了验证RNA-Seq数据的可靠性,采用qRT-PCR方法检测15个DEGs的表达水平,包括6个莽草酸代谢相关的DEGs、4个共表达模块筛选到的关键基因和5个随机挑选的DEGs(图11)。结果表明,qRT-PCR和RNA-Seq的基因表达变化趋势基本一致,二者数据结果呈正相关,表明本研究中基于转录组数据得到的DEGs是可信的。

A:DEGs富集到的植物激素信号通路图;B:热图表示与植物激素信号转导相关DEGs的表达水平

A:样本聚类树;B:无尺度容适曲线及平均连通度曲线;C:基因聚类树及模块切割,基因聚类树的每一个分支对应一个模块
3 讨论
3.1 草甘膦能够干扰茶树叶片莽草酸代谢
莽草酸作为草甘膦抑制莽草酸代谢的积累产物,其含量是最能反映草甘膦毒性的指标之一[24]。在草甘膦处理3 d内,茶树叶片中的莽草酸含量低于处理前,而在7 d时莽草酸含量大量积累,表明茶树莽草酸代谢在试验处理3 d后明显受到了草甘膦的抑制。前人研究表明,莽草酸途径上的关键酶基因和在抵御茶树逆境胁迫中具有重要作用[25-26],此外,作为草甘膦胁迫的作用靶点酶基因,已被广泛用于转基因抗草甘膦作物的开发[27]。本研究中()和()在处理不同时间点的表达量上调表达,说明它们可能参与茶树对草甘膦胁迫的抵御反应,在草甘膦处理前期保护茶树以免莽草酸积累中毒。
3.2 草甘膦影响茶树苯丙烷、类黄酮生物合成及激素信号转导途径相关基因的转录
草甘膦干扰植物莽草酸代谢的同时,也会影响以芳香族氨基酸为前体物质的次生代谢物(如植物激素、类黄酮)合成[28]。ZHAI等[29]研究表明草甘膦诱导了水稻谷胱甘肽代谢、介导激素信号通路相关基因的差异表达。MESNAGE等[30]认为草甘膦能够干扰土壤真菌的蛋白合成、次生代谢及应激反应,导致土壤质量下降。课题组前期发现,草甘膦处理显著改变了茶树叶片中游离氨基酸、儿茶素和生物碱总量及组分构成[12]。本研究中草甘膦处理不同时期(0.25、1、3和7 d)与处理前(0 d)比对的差异基因均显著富集于苯丙烷、类黄酮生物合成途径及植物激素信号转导途径。说明草甘膦干扰茶树莽草酸代谢的同时,也会诱导下游代谢途径苯丙烷、类黄酮生物合成及激素信号转导相关基因的差异表达。此外,苯丙烷代谢途径是参与植物防御反应的主要次级代谢通路之一,苯丙烷途径相关基因大量表达有利于加速下游分支代谢产物(如类黄酮、花青素)的生成[31-32]。本研究中参与茶树苯丙烷(、、和)及类黄酮(、、、和)生物合成途径的多个关键基因在草甘膦处理0.25 d表达量下调,说明草甘膦在处理能够迅速干扰茶树苯丙烷、类黄酮化合物的生物合成,在前期对茶树次生代谢产物的合成产生不利影响。

横坐标代表不同表型,纵坐标代表不同模块。图中左侧一列数字表示该模块的基因数目,右侧每组数据表示模块与表型的相关性系数r值及显著性P值(括号内)。红色代表模块与表型的相关性较大,蓝色代表模块与表型的相关性较小
3.3 基于WGCNA鉴定茶树响应草甘膦相关的共表达基因
利用WGCNA分析法,可特异地筛选出与目标性状相关的基因,并进行模块化分类,得到具有高度生物学意义的共表达模块[33]。莽草酸含量可直接反映植物对草甘膦的响应情况[24]。本研究利用WGCNA法将转录组与莽草酸含量数据相关联,并对模块进行KEGG功能富集分析,最终筛选到2个与草甘膦响应高度相关的关键模块,分别是green模块(2 024个基因)和brown模块(2 305个基因)。WGCNA共表达分析中连通度高的基因通常起重要调控作用[33]。ZHAO等[34]认为草甘膦诱导了蜜蜂许多农药解毒和抗性基因上调表达,以防御草甘膦对蜜蜂生长发育产生不利影响。本研究中、和在草甘膦处理7 d明显上调表达(图10),说明、和可能在草甘膦处理后期降低草甘膦对茶树的不利作用。DOGRAMACI等[35]研究表明杂草通过细胞转运、激素信号传导和解毒机制等相互作用,以防御草甘膦诱导的莽草酸积累中毒。JIANG等[6]发现大豆转运蛋白基因能够降低草甘膦除草剂毒性作用。由此可知,、和等可能通过参与离子运输、膜运输和激素信号转导等相互作用,在茶树抵御草甘膦逆境过程中发挥重要作用。此外,(乙烯响应因子)是模块中筛选出响应草甘膦的关键基因之一,本研究中乙烯信号途径下游基因()在处理不同时间点表达水平较高(87.88<TPM<271.03),并上调表达。因此,在茶树响应草甘膦胁迫的激素信号转导中,乙烯可能起重要作用。

纵轴表示pathway名称,横轴表示Rich factor;点的大小表示此pathway中基因个数多少,点的颜色对应于不同的P值范围。图中仅展示P-value<0.05的前提下富集程度在前20的KEGG Pathway

三角形节点为连通性排名前3的基因,黑色边框节点为转录因子基因
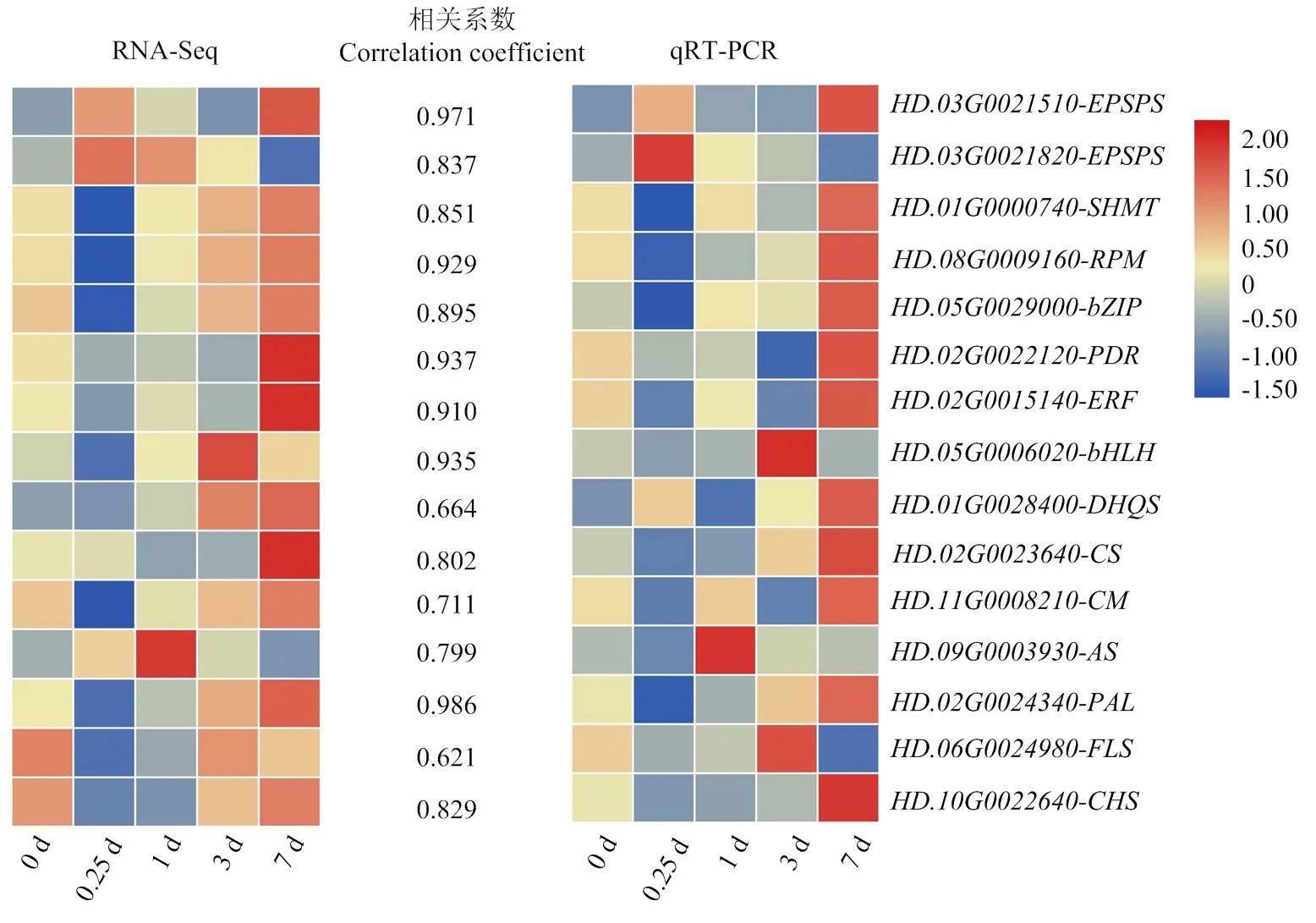
热图分别表示RNA-Seq和qRT-PCR的基因表达水平;两个热图之间的值代表每个基因qRT-PCR和RNA-seq值的相关系数
4 结论
本研究利用草甘膦处理后茶树叶片的RNA-seq及莽草酸表型数据,发现草甘膦能够干扰茶树叶片中莽草酸代谢,并诱导其下游苯丙烷、类黄酮生物合成及植物激素信号转导途径相关基因的差异表达,而乙烯可能在茶树响应草甘膦胁迫的激素信号转导中起重要作用。通过WGCNA方法发现2个与草甘膦响应高度相关的关键基因模块,共表达分析发现6个关键调控基因(、、和)在茶树抵御草甘膦逆境过程中发挥重要作用。
[1] 王鹏杰, 岳川, 陈笛, 郑玉成, 郑知临, 林浥, 杨江帆, 叶乃兴. 茶树CsWRKY6、CsWRKY31和CsWRKY48基因的分离及表达分析. 浙江大学学报(农业与生命科学版), 2019, 45(1): 30-38.
WANG P J, YUE C, CHEN D, ZHENG Y C, ZHENG Z L, LIN Y, YANG J F, YE N X. Isolation and expression analysis of CsWRKY6, CsWRKY31 and CsWRKY48 genes in tea plant. Journal of Zhejiang University (Agriculture and Life Sciences), 2019, 45(1): 30-38. (in Chinese)
[2] 唐杏燕, 邵增琅, 杨路成, 裴少芬, 岳鹏翔, 王晓霞. 茶园中草甘膦在靶标杂草和非靶标茶树中的吸收、转运、分布和代谢. 食品安全质量检测学报, 2018, 9(18): 140-145.
Tang X Y, Shao Z L, Yang L C, PEI S F, YUE P X, WANG X X. Uptake, translocation, distribution and metabolism of glyphosate in target weeds and non-target tea trees in tea garden. Journal of Food Safety Quality Inspection, 2018, 9(18): 140-145. (in Chinese)
[3] 杨亚琴, 冯书惠, 胡永建, 李圆圆, 王会锋, 刘进玺, 钟红舰. 气相色谱-质谱法测定绿茶中草甘膦和氨甲基膦酸残留量. 茶叶科学, 2020, 40(1): 125-132.
Yang Y Q, Feng S H, Hu Y J, LI Y Y, WANG H F, LIU J X, ZHONG H J. Determination of glyphosate and aminomethylphosphonic acid residue in green tea by gas chromatography-mass spectrometry. Journal of Tea Science, 2020, 40(1): 125-132. (in Chinese)
[4] 郭永春, 陈金发, 赵峰, 王淑燕, 王鹏杰, 周鹏, 欧阳立群, 金珊, 叶乃兴. 草甘膦及其代谢物氨甲基膦酸在茶树体中的分布研究. 茶叶科学, 2020, 40(4): 510-518.
GUO Y C, CHEN J F, ZHAO F, WANG S Y, WANG P J, ZHOU P, OUYANG L Q, JIN S, YE N X. Study on the distribution of glyphosate and its metabolite aminomethylphosphonic acid inJournal of Tea Science, 2020, 40(4): 510-518. (in Chinese)
[5] 于惠林, 贾芳, 全宗华, 崔海兰, 李香菊. 施用草甘膦对转基因抗除草剂大豆田杂草防除、大豆安全性及杂草发生的影响. 中国农业科学, 2020, 53(6): 1166-1177.
YU H L, JIA F, QUAN Z H, CUI H L, LI X J. Effects of glyphosate on weed control, soybean safety and weed occurrence in transgenic herbicide-resistant soybean. Scientia Agricultura Sinica, 2020, 53(6): 1166-1177. (in Chinese)
[6] JIANG L X, JIN L G, GUO Y, TAO B, QIU L J. Glyphosate effects on the gene expression of the apical bud in soybean (). Biochemical and Biophysical Research Communications, 2013, 437(4): 544-549.
[7] RAINIO M J, MARGUS A, VIRTANEN V, LINDSTRÖM L, SALMINEN J P, SAIKKONEN K, HELANDER M. Glyphosate- based herbicide has soil-mediated effects on potato glycoalkaloids and oxidative status of a potato pest. Chemosphere, 2020, 258: 127254.
[8] MALAGODA M, OHM J, HOWATT K A, GREEN A, SIMSEK S. Effects of pre-harvest glyphosate use on protein composition and shikimic acid accumulation in spring wheat. Food Chemistry, 2020, 332: 127422.
[9] Tong M M, Gao W J, Jiao W, Hou R Y. Uptake, translocation, metabolism, and distribution of glyphosate in nontarget tea plant (L.). Journal of Agricultural and Food Chemistry, 2017, 65(35): 7638-7646.
[10] 高万君, 张永志, 童蒙蒙, 马慧勤, 钱珊珊, 王天雨, 李叶云, 吴慧平, 侯如燕. 茶园常用除草剂田间药效试验与残留动态. 茶叶科学, 2019, 39(5): 587-594.
Gao W J, Zhang Y Z, Tong M M, Ma H Q, Qian S S, Wang T Y, Li Y Y, Wu H P, Hou R Y. Weeds control effect and residues of several herbicides in tea gardens. Journal of Tea Science, 2019, 39(5): 587-594. (in Chinese)
[11] 高万君, 李叶云, 侯如燕. 茶叶中草甘膦残留现状与对策. 中国茶叶, 2021, 43(4): 20-24.
Gao W J, Li Y Y, Hou R Y. Status and countermeasures of glyphosate residues in tea. China Tea, 2021, 43(4): 20-24. (in Chinese)
[12] 郭永春, 王淑燕, 王鹏杰, 陈金发, 周鹏, 欧阳立群, 赵峰, 叶乃兴. 草甘膦对茶树叶片主要生化成分的影响. 天然产物研究与开发, 2021, 33(3): 394-401.
GUO Y C, WANG S Y, WANG P J, CHEN J F, ZHOU P, OUYANG L Q, ZHAO F, YE N X. The effect of glyphosate on the main biochemical components of tea leaves. Natural Product Research and Development, 2021, 33(3): 394-401. (in Chinese)
[13] WANG P J, YU J X, JIN S, CHEN S, YUE C, WANG W L, GAO S L, CAO H L, ZHENG Y C, GU M Y, CHEN X J, SUN Y, GUO Y Q, YANG J F, ZHANG X T, YE N X. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome. Horticulture Research, 2021, 8(1): 107.
[14] TRAPNELL C, ROBERTS A, GOFF L, PERTEA G, KIM D, KELLEY D R, PIMENTEL H, SALZBERG S L, RINN J L, PACHTER L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 2012, 7(3): 562-578.
[15] LI B, DEWEY C N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 2011, 12: 323.
[16] ROBINSON M D, MCCARTHY D J, SMYTH G K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 2010, 26(1): 139-140.
[17] XIE C, MAO X Z, HUANG J J, DING Y, WU J M, DONG S, KONG L, GAO G, LI C Y, WEI L P. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research, 2011, 39: W316-W322.
[18] CHEN C, XIA R, CHEN H, XIA T. TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. BioRxiv, 2018.
[19] LANGFELDER P, HORVATH S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 2008, 9: 559.
[20] KOHL M, WIESE S, WARSCHEID B. Cytoscape: Software for visualization and analysis of biological networks. Methods in Molecular Biology, 2011, 696: 291-303.
[21] 王鹏杰, 曹红利, 陈丹, 陈笛, 陈桂信, 杨江帆, 叶乃兴. 茶树脂肪酸去饱和酶家族基因的克隆与表达分析. 园艺学报, 2020, 47(6): 1141-1152.
WANG P J, CAO H L, CHEN D, CHEN D, CHEN G X, YANG J F, YE N X. Cloning and expression analysis of fatty acid desaturase family genes in. Acta Horticulturae Sinica, 2020, 47(6): 1141-1152. (in Chinese)
[22] WU Z J, TIAN C, JIANG Q A, LI X H, ZHUANG J. Selection of suitable reference genes for qRT-PCR normalization during leaf development and hormonal stimuli in tea plant (). Scientific Reports, 2016, 6: 19748.
[23] LIVAK K J, SCHMITTGEN T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods, 2001, 25: 402-408.
[24] ZELAYA I A, ANDERSON J A H, OWEN M D K, LANDES R D. Evaluation of spectrophotometric and HPLC methods for shikimic acid determination in plants: Models in glyphosate-resistant and -susceptible crops. Journal of Agricultural and Food Chemistry, 2011, 59(6): 2202-2212.
[25] 郭永春, 王鹏杰, 谷梦雅, 王淑燕, 赵峰, 叶乃兴. 茶树5-烯醇式丙酮酰莽草酸-3-磷酸合成酶基因的克隆及表达. 应用与环境生物学报, 2020. https://doi.org/10.19675/j.cnki.1006-687X.2020.10031.
GUO Y C, WANG P J, GU M Y, WANG S Y, ZHAO F, YE N X. Cloning and expression of 5-enolpyruvylshikimate-3-phosphate synthase gene from tea plants. Chinese Journal of Applied and Environmental Biology, 2020. https://doi.org/10.19675/j.cnki.1006- 687X.2020.10031. (in Chinese)
[26] 孙平, 章国营, 向萍, 林金科, 赖钟雄. 茶树中莽草酸途径DHD/SDH基因的表达调控. 应用与环境生物学报, 2018, 24(2): 322-327.
SUN P, ZHANG G Y, XIANG P, LIN J K, LAI Z X. Expression and regulation of the shikimic acid pathway gene DHD/SDH in tea plant (). Chinese Journal of Applied and Environmental Biology, 2018, 24(2): 322-327. (in Chinese)
[27] ACHARY V M M, SHERI V, MANNA M, PANDITI V, BORPHUKAN B, RAM B, AGARWAL A, FARTYAL D, TEOTIA D, MASAKAPALLI S K, AGRAWAL P K, REDDY M K. Overexpression of improved EPSPS gene results in field level glyphosate tolerance and higher grain yield in rice. Plant Biotechnology Journal, 2020, 18(12): 2504-2519.
[28] 陈延儒. 采后硝普钠处理对苹果果实品质、莽草酸和苯丙烷代谢途径的影响[D]. 锦州: 渤海大学, 2019.
CHEN Y R. Effect of postharvest sodium nitroprusside treatment on the quality, shikimate and phenylpropanoid pathway of apple fruit [D]. Jinzhou: Bohai University, 2019. (in Chinese)
[29] ZHAI R R, YE S H, ZHU G F, LU Y T, YE J, YU F M, CHU Q R, ZHANG X M. Identification and integrated analysis of glyphosate stress-responsive microRNAs, lncRNAs, and mRNAs in rice using genome-wide high-throughput sequencing. BMC Genomics, 2020, 21(1): 238.
[30] MESNAGE R, OESTREICHER N, POIRIER F, NICOLAS V, BOURSIER C, VÉLOT C. Transcriptome profiling of the fungusexposed to a commercial glyphosate-based herbicide under conditions of apparent herbicide tolerance. Environmental Research, 2020, 182: 109116.
[31] MENG J, WANG B, HE G, WANG Y, TANG X F, WANG S M, MA Y B, FU C X, CHAI G H, ZHOU G K. Metabolomics integrated with transcriptomics reveals redirection of the phenylpropanoids metabolic flux in. Journal of Agricultural and Food Chemistry, 2019, 67(11): 3284-3291.
[32] DONG N Q, LIN H X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. Journal of Integrative Plant Biology, 2021, 63(1): 180-209.
[33] 秦天元, 孙超, 毕真真, 梁文君, 李鹏程, 张俊莲, 白江平. 基于WGCNA的马铃薯根系抗旱相关共表达模块鉴定和核心基因发掘. 作物学报, 2020, 46(7): 1033-1051.
QIN T Y, SUN C, BI Z Z, LIANG W J, LI P C, ZHANG J L, BAI J P. Identification of drought-related co-expression modules and hub genes in potato roots based on WGCNA. Acta Agronomica Sinica, 2020, 46(7): 1033-1051. (in Chinese)
[34] ZHAO H, LI G L, GUO D Z, WANG Y, LIU Q X, GAO Z, WANG H F, LIU Z G, GUO X Q, XU B H. Transcriptomic and metabolomic landscape of the molecular effects of glyphosate commercial formulation onmellifera ligustica andcerana. The Science of the Total Environment, 2020, 744: 140819.
[35] DOGRAMACI M, ANDERSON J V, CHAO W S, HORVATH D P, HERNANDEZ A G, MIKEL M A, FOLEY M E. Foliar glyphosate treatment alters transcript and hormone profiles in crown buds of leafy spurge and induces dwarfed and bushy phenotypes throughout its perennial lifecycle. The Plant Genome, 2017, 10(3): 98.
附表1 转录组数据RT-qPCR 验证引物序列
Table S1 Transcriptome data RT-qPCR validation primer sequences

Identification of Co-Expression Gene Related to Tea Plant Response to Glyphosate Based on WGCNA

1College of Horticulture, Fujian Agriculture and Forestry University/Key Laboratory of Tea Science in Universities of Fujian Province, Fuzhou 350002;2School of Pharmacy, Fujian University of Chinese Medicine, Fuzhou 350122
【】This study aimed at analyzing both expression patterns and regulatory pathways of tea plants in response to glyphosate stressing, which could revealed the effect of glyphosate herbicides on tea plants at transcriptional level and identify key genes of tea plants. 【】cv Jin-guanyin was applied as material plant. A recommended concentration of glyphosate was irrigated to test plants. The leave samples were collected at different time intervals (0, 0.25, 1, 3 and 7 d). The samples were sequenced by transcriptome, the content of shikimic acid was also quantified.The WGCNA method was used to jointly analyze transcriptome and shikimic acid content data, to identify co-expressed gene modules related to glyphosate response, and to screen out key regulatory genes. 【】The content of shikimic acid in tea leaves reduced gradually during first 3 days. However, it suddenly reached a peak on the 7th day (6.99 times compared with no glyphosate treated sample). A total of 12 568 differential expression genes (DEGs) were also identified, which mainly enriched in phenylpropane, flavonoid biosynthesis and plant hormone signal transduction pathways. In addition, the glyphosate treatment induced 24, 52, 31 and 69 genes respectively which related to shikimic acid metabolism, phenylpropane, flavonoid biosynthesis and hormone signal transduction pathways. A total of 19 modules were screened out by WGCNA method. The correlation analysis of transcriptome and shikimic acid content indicated two key modules, including 2 024 and 2 305 genes, respectively. The top 50 genes with the highest connectivity in the key modules were selected for co-expression analysis, and 6 key response genes were obtained, including 2 resistance genes (and), 1 drug resistance gene (), 1 ion transport gene (), 1 membrane transport gene (), and 1 transcription factor gene ().【】Glyphosate could affect downstream genes transcription of phenylpropane, flavonoid biosynthesis and hormone signal transduction pathways by interfering shikimic acid metabolism of tea plants. In addition, this study also identified two co-expression modules closely related to glyphosate response, and found that multiple potential candidate genes and transcription factors could resist glyphosate stress, such as,,,,and
; glyphosate; shikimic acid; transcriptome; WGCNA

10.3864/j.issn.0578-1752.2022.01.013
2021-03-12;
2021-05-10
福建省“2011协同创新中心”中国乌龙茶产业协同创新中心专项(闽教科[2015]75号)、福建农林大学科技创新专项基金(CXZX2017181)、福建农林大学茶产业链科技创新与服务体系建设项目(2020A01)、福建农林大学园艺学院优秀硕士学位论文资助基金(2019S01)
郭永春,E-mail:1062941682@qq.com。通信作者叶乃兴,E-mail:ynxtea@126.com。通信作者赵峰,E-mail:zhaofeng0591@fjtcm.edu.cn
(责任编辑 赵伶俐)
