Fractalkine通过激活p38MAPK信号通路调控狼疮性肾炎小鼠Treg细胞凋亡①
2022-02-10马敬雪巩奇明潘秀虹何林檩尤燕舞右江民族医学院附属医院肾内科百色533000
马敬雪 巩奇明 潘秀虹 何林檩 尤燕舞 (右江民族医学院附属医院肾内科,百色 533000)
目前认为,狼疮性肾炎(lupus nephritis,LN)的 发生是由于机体的免疫应答系统出现异常,导致抗原抗体免疫复合物沉积于肾脏,随之产生趋化因子、黏附因子、促炎症因子等细胞因子的释放,引起肾脏组织发生氧化应激和炎症反应,最终导致肾脏系膜增生和内皮细胞损伤。调节性T细胞(T regulatory cells,Treg)是一类调控机体免疫功能的新型T细胞亚群,能维持免疫系统对自身成分的耐受,使机体保持免疫稳态。当其功能或数量发生异常变化时,会导致多种自身免疫性疾病的发生。Treg免疫疗法有可能成为治疗自身免疫性疾病的新途径。Fractalkine(FKN)是CX3C-类趋化因子超家族中的唯一成员[1]。近年的研究证实,MRL/lpr小鼠肾皮质FKN的表达量较正常对照组明显升高[2]。同样,有学者在临床研究中发现,与健康者对比,SLE患者血清中FKN的水平明显升高,并且与疾病的活动度呈正相关,与外周血的Treg细胞计数呈负相关[3]。另一项研究显示,在发病前或LN早期,输注了FKN拮抗剂的MRL/lpr小鼠,其肾小球细胞增生、肾小球硬化、新月体形成及血管炎等表现明显减轻,表明FKN拮抗剂能延缓LN的发病并改善病情进展[4]。p38MAPK可被趋化因子、炎症因子和其他外部应激激活。p38MAPK信号通路一旦被激活,就可以启动p53、PAX6、CREB和ATF2等转录因子的产生,从而调控促炎症或凋亡相关基因的产生[5]。p38MAPK信号通路大量存在于T细胞和B细胞中,并且参与T细胞和B细胞的许多细胞过程和反应[6-9]。许多数据表明,p38MAPK活性在炎症反应、正常免疫过程中起关键作用[10]。此外,p38MAPK被激活并参与人类自身免疫性疾病的发病机制,如类风湿关节炎和系统性红斑狼疮[11-12]。既往的研究发现,小鼠系膜细胞的增殖受FKN的影响,在高浓度FKN条件下,p38MAPK信号通路被激活。另外李波[13]研究证实,在单个核细胞中FKN可激活p38MAPK信号转导通路,从而调节单个核细胞的功能。然而迄今为止,FKN是否可通过p38MAPK信号通路参与Treg细胞功能的调节鲜有报道。本研究旨在构建LN小鼠模型,探究FKN在LN小鼠肾组织的表达及在LN Treg细胞凋亡中的作用及其分子机制,为阐明LN的发病机制提供了实验依据。
1 材料与方法
1.1 材料 8周龄的BALB/c雌性小鼠80只,购自湖南长沙天勤实验动物中心,动物许可证号:SCXK(湘)2019-0014;Pristane(S44062,上海源业生物科技有限公司);FKN中和抗体(MAB571,R&D Systems,USA);用于动物的SB203580(S1076,Selleck,China);用于动物的U-46619(16450,Cayman,China);IL-2、TGF-β(中国新生物科学公司);用于细胞的U-46619(Santa Cruz Biotechnology,USA);用于细胞的SB203580(S1863-1,Beyotime Biotechnology,China);胰蛋白酶、双抗、PBS、BCA蛋白定量试剂盒(上海碧云天生物技术有限公司);Annexin V-FITC、PI凋亡试剂检测盒(BD);胎牛血清、RPMI1640、红细胞裂解液、Hanks液(Gibco);免疫磁珠分选试剂盒(CD4+CD25+Regulatory T Cell Ⅰ solation Kit,No.130-091-014,Miltenyi Biotec);兔抗 Bax一抗、兔抗Bcl-2一抗、鼠抗Cyt-c一抗、兔抗FKN一抗、兔抗Foxp3一抗、兔抗p38一抗(Abcam);兔抗phosphop38一抗(Affinity公司);鼠抗GAPDH(Proteintech);山羊抗鼠IgG/辣根酶标记及山羊抗兔IgG/辣根酶标记(中杉金桥);SiRNA腺病毒株(上海吉凯公司)。
1.2 方法
1.2.1 动物模型构建及分组 雌性BALB/c小鼠一次性腹腔注射500 µl Pristane,喂养期间小鼠自由饮水及进食。12周后,根据课题组前期的验证方法检测24 h尿蛋白含量、血清抗核抗体表达及PASM染色观察肾组织病理改变以鉴定LN小鼠模型成功建立[14]。将实验小鼠随机分为正常组、Anti-FKN组、SB203580组、U-46619组、模型组、模型+Anti-FKN组、模型+SB203580组、模型+U-46619组、模型+Anti-FKN+SB203580组及模型+Anti-FKN+U-46619组,每组5只小鼠。各组小鼠腹腔注射给药:①正常组:给予正常小鼠腹腔注射生理盐水500 µl/(只·d),连续2周;②Anti-FKN组:给予正常小鼠腹腔注射Anti-FKN 5.0 µg/(只·d),连续2周;③SB203580组:给予正常小鼠腹腔注射SB203580 2 mg/(kg·d),连续2周;④U-46619组:给予正常小鼠腹腔注射U-46619 2 mg/(kg·d),连续2周;⑤模型组:给予模型组小鼠腹腔注射生理盐水500 µl/(只·d),连续12周;⑥模型+Anti-FKN组:给予模型小鼠腹腔注射Anti-FKN 5.0 µg/(只·d),连续2周;⑦模型+SB203580组:给予模型小鼠腹腔注射SB203580 2 mg/(kg·d),连续2周;⑧模型+U-46619组:给予模型小鼠腹腔注射U-46619 2 mg/(kg·d),连续2周;⑨模型+Anti-FKN+SB203580组:给予模型小鼠腹腔注射Anti-FKN 5.0 µg/(只·d)及SB203580 2 mg/(kg·d),连续2周;⑩模型+Anti-FKN+U-46619组:给予模型小鼠腹腔注射Anti-FKN 5.0 µg/(只·d)及U-46619 2 mg/(kg·d),连续2周。干预2周后,采用颈椎脱臼法处死小鼠,摘取肾脏。
1.2.2 免疫组化方法 肾组织石蜡切片进行程序性脱蜡,滴加一抗工作液4 ℃过夜,PBS缓冲液清洗后滴加二抗37 ℃孵育30 min,用二氨基联苯胺显色,然后用苏木素复染。显微镜下观察肾组织中的FKN、p-p38、Foxp3的蛋白表达情况。
1.2.3 细胞分选及培养 处死LN模型小鼠后浸泡在75%无水乙醇10 min,取出脾脏剪碎后置于200目的筛网过滤网中,用注射器的活塞进行研磨(研磨过程中不断加入Hank's液,保持脾脏的湿度)滤出单个细胞悬液;离心后加入等量红细胞裂解液,放入37 ℃摇床5 min,制备RPMI1640培养基脾细胞悬液后转移到培养瓶中,在37 ℃、5%CO2培养箱中培养3~4 h,使贴壁细胞贴壁;用巴氏滴管轻轻吸出悬浮细胞(即总淋巴细胞),并计数;加入终浓度为10 ng/ml的TGF-β和IL-2促进细胞增殖及分化,培养42 h后开始分选;收集细胞并计数,每1×107个细胞用40 µl 分选Buffer重悬;每1×107个细胞加入10 µl CD4+CD25+Regulatory T cell BIoTin-Antibody Cocktail,2~8 ℃孵育10 min;每1×107个细胞体系中,加入38 µl 分选Buffer,20 µl Anti-Biotin Microbeads,2 µl CD25-PE Antibody,混匀后 2~8 ℃孵育 15 min;过柱收集不吸附的液体(即CD4+Treg细胞);液体离心(300 g,5 min),用90 µl分选Buffer重悬,加入10 µl Anti-PE Microbeads,混 匀 后 2~8 ℃ 孵 育 15 min;过柱收集吸附于磁柱上的磁珠细胞(即CD4+CD25+Treg细胞)。收集分选出来的CD4+CD25+Treg细胞转移到RPMI1640完全培养基中,在37 ℃、5%CO2培养箱中培养。
1.2.4 细胞转染及分组 按每孔2×105个/ml在6孔板接种Treg细胞,常规培养至70%融合时更换无血清培养液,将阳性及阴性载体腺病毒加入无血清无抗生素的培养基中并充分混匀,使病毒终浓度为6×107pfu/ml,于室温下孵育5 min后转移至37 ℃、5%CO2条件下培养,转染48 h后于荧光显微镜下观察转染情况。转染成功后24~48 h观察细胞状态,生长状态良好时,加入8 µg/ml的嘌呤霉素(Puromycin)选择转染细胞,直至正常细胞完全死亡,得到稳定转染细胞系。并采用Western blot法鉴定转染结果。将LN Treg细胞分为7组:对照组、空载体组、FKN-KD组、U-46619组、SB203580组、FKN-KD+U-46619组及FKN-KD+SB203580组。在37 ℃、5%CO2饱和湿度条件下继续培养。培养24 h后收集各组细胞进行后续实验。
1.2.5 Western blot法 收集各组细胞至离心管离心,弃上清后用预冷的PBS液洗2次,加入150 µl预冷PI裂解液,冰浴30 min,4 ℃、12 000 r/min离心10 min,取上清,以二喹啉甲酸(BCA)法测定提取蛋白质量浓度。每个样本取30~50 µg蛋白加入5×SDS上样缓冲液煮沸变性后,SDS-PAGE电泳,湿式电印迹法300 mA恒流转至PVDF膜,3%BSA室温封闭1 h,加TBST洗膜缓冲液洗3次,每次10 min加入一抗孵育4 ℃过夜,加TBST洗膜缓冲液洗3次,每次10 min,加入HRP标记的羊抗兔或羊抗小鼠IgG(H+L),常温振荡孵育1 h,洗涤后采用Chemi-DocXRS 凝胶成像系统Qunatity One软件采集图像,灰度值采用NIH Image软件(National Institute of-Health,Bethesda,Md,USA)测定。以目的蛋白条带与GAPDH蛋白条带灰度值的比值表示其相对含量。重复试验3次。
1.3 统计学处理 采用SPSS23.0统计软件进行数据分析,所有数据均进行正态分布和方差齐性检验。服从正态分布的数据以±s表示,两组间比较采用独立样本t检验,多组间采用单因素方差分析。非正态分布则以中位数和极值表示,采用秩和检验。每个实验组重复3次。P<0.05为差异有统计学意义。
2 结果
2.1 FKN、 p38MAPK及Foxp3在LN小鼠肾组织中的表达 经U-46619干预后的正常小鼠及模型小鼠均在5~7 d内连续死亡。与正常对照组相比,模型组小鼠肾组织中的FKN及磷酸化p38MAPK的表达明显上调(P<0.05),Foxp3染色阳性的Treg细胞数明显减少(P<0.05);与模型组相比,模型+anti-FKN组和模型+SB203580组肾组织中的FKN及磷酸化p38MAPK的表达明显下调(P<0.05),Foxp3染色阳性的Treg细胞数明显增加(P<0.05);与模型+anti-FKN组和模型+SB203580组相比,模型+anti-FKN+SB203580组肾组织中的FKN及磷酸化p38MAPK的表达进一步下调(P<0.05),Foxp3染色阳性的Treg细胞数明显增加(P<0.05)。然而,模型+anti-FKN组和模型+SB203580组肾组织中的FKN及磷酸化p38MAPK的表达差异无统计学意义(P>0.05,图1)。

图1 FKN、p38MAPK及Foxp3在LN小鼠肾组织中的表达Fig.1 Expression of FKN , p38MAPK and Foxp3 in kidney of mice with LN
2.2 细胞转染结果的鉴定 根据腺病毒转染细胞的操作说明书转染Treg细胞24 h后用荧光倒置显微镜观察,转染组细胞可见绿色荧光均匀分布于悬浮Treg细胞内(图2)。

图2 转染后Treg细胞绿色荧光蛋白表达情况Fig.2 Expression of green fluorescent protein in transfected Treg cells
收集细胞并提取总蛋白,采用Western blot检测正常Treg细胞及敲低FKN的Treg细胞中FKN的表达量。结果显示,两组细胞中均有FKN的表达,转染组细胞中的FKN表达量明显低于对照组(P<0.05,图3)。

图3 敲低FKN基因后Treg细胞中FKN蛋白表达变化Fig.3 Expression of FKN protein in Tregs cells with FKN knockdown
2.3 FKN通过激活LN Treg细胞中p38MAPK磷酸化抑制Foxp3的表达 与对照组相比,敲低FKN后p38MAPK的磷酸化水平明显降低,而Foxp3表达明显增加,差异均有统计学意义(P<0.01)。与FKNKD组相比,FKN-KD+U-46619组的p38MAPK磷酸化水平明显升高,而Foxp3表达明显降低,FKN-KD+SB203580组的p38MAPK磷酸化水平进一步降低,而Foxp3表达进一步增加,差异均有统计学意义(P<0.01),然而,SB203580组的p38MAPK磷酸化水平和Foxp3表达无明显变化,差异无统计学意义(P>0.05)。与对照组相比,U-46619组的p38MAPK磷酸化水平进一步升高,而Foxp3表达进一步降低;SB203580组的p38MAPK磷酸化水平明显降低,而Foxp3表达明显增加,差异均有统计学意义(P<0.01),FKN-KD+U-46619组中的p38MAPK磷酸化和Foxp3的表达水平均无明显变化,差异无统计学意义(P>0.05)。然而,非磷酸化p38MAPK均无明显改变,差异无统计学意义(P>0.05,图4)。
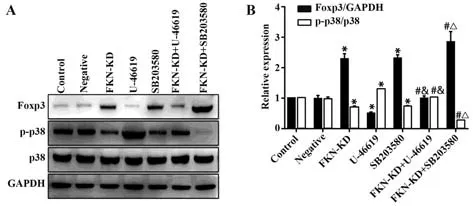
图4 Western blot检 测 各 组 Foxp3、p-p38MAPK、p38MAPK相关蛋白的表达水平Fig.4 Expression levels of Foxp3, p-p38MAPK, p38MAPK protein detected by Western blot
2.4 FKN及p38MAPK信号通路影响相关凋亡蛋白的表达 与对照组相比,FKN-KD组、SB203580组中的Bax及Cyt-c的表达明显减少,Bcl-2的表达明显增加,U-46619组中的Bax及Cyt-c的表达明显增加,Bcl-2的表达明显减少,差异均有统计学意义(P<0.01)。与FKN-KD组相比,FKN-KD+U-46619组中Bax及Cyt-c的表达明显增加,Bcl-2的表达明显减少,而FKN-KD+SB203580组中Bax及Cyt-c的表达明显减少,Bcl-2的表达明显增加,差异均有统计学意义(P<0.01)。与 U-46619组相比,FKN-KD+U-46619组中Bax及Cyt-c的表达明显减少,Bcl-2的表达明显增加,差异有统计学意义(P<0.01)。与SB203580组相比,FKN-KD+SB203580组中Bax及Cyt-c的表达明显减少,Bcl-2的表达明显增加,差异均有统计学意义(P<0.01)。然而,与对照组相比,FKN-KD+U-46619组的Bax、Bcl-2及Cyt-c的表达均无明显变化,差异无统计学意义(P>0.05)。与FKN-KD组相比,SB203580组的Bax、Bcl-2及Cyt-c的表达均无明显变化,差异无统计学意义(P>0.05,图5)。
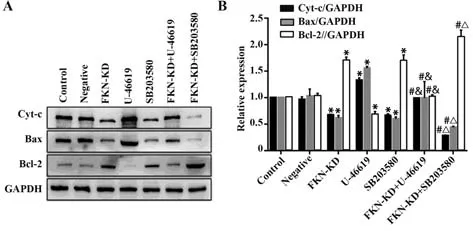
图5 Western blot检测各组Bax、Bcl-2、Cyt-c相关蛋白的表达水平Fig.5 Expression levels of Bax, Bcl-2, Cyt-c protein detected by Western blot
3 讨论
LN是一种自身免疫性疾病,其特征在于T细胞功能障碍,包括Treg细胞的凋亡及损伤[15-16]。Treg细胞具有免疫抑制和维持免疫耐受的功能,它在免疫调节和发育耐受中起着关键性的作用[15,17-19]。近年来,研究发现FKN参与细胞黏附、生长调节和炎症免疫反应[20]。FKN在类风湿关节炎、系统性红斑狼疮、多发性硬化和肿瘤等免疫炎症疾病组织中高表达,并且与疾病活动性有关[21-27]。此外,FKN在LN患者的血清中高表达,并且与LN患者的肾损害程度呈正相关[4,22]。研究证实,FKN是一种自身免疫性疾病中T细胞迁移相关的关键趋化因子。FKN在Treg细胞中过度表达,对Treg细胞具有较强的趋化作用,从而影响Treg细胞的功能,最终导致免疫失衡,加速免疫疾病的进展[28]。因此,本课题组认为趋化因子FKN调控Treg细胞的功能,从而参与LN的发生发展过程。重要的是,本课题组的前期研究已经证实,在LN小鼠模型的血液中Treg细胞的比例显著降低,肾组织中Foxp3表达量也显著减少[14]。而在本研究中,采用免疫组化法、Western blot法检测发现,抑制和(或)敲低FKN的表达后,则可增加LN肾组织及Treg细胞中Foxp3的表达,减少Treg细胞中促凋亡蛋白的表达,增加抗凋亡蛋白的表达[29]。然而,FKN如何调控Foxp3的表达及诱导Treg细胞凋亡的分子机制至今仍未阐明。
p38MAPK家族的四个主要成员包括p38、MAPK11、SAPK4和SAPK3,它们被趋化因子、炎症因子、紫外线辐射、热休克、高渗压和其他外部应激激活。p38MAPK通路一旦被激活,就可以启动p53、PAX6、CREB和ATF2等转录因子的产生,从而调控促炎症或凋亡相关基因的产生[5]。p38MAPK介导重要生物学反应的信号,包括转录因子的磷酸化、炎症细胞因子和趋化因子的产生[30-31]。LIU等[32]发现,单核细胞中的p38MAPK信号通路在LN中起重要的致病作用。另一个研究发现,SLE小鼠的肾脏组织中p38MAPK mRNA水平显著升高[12]。系统性红斑狼疮小鼠的肾损伤和IgG的产生依赖于p38MAPK的激活,p38MAPK的激活上调了SLE小鼠的趋化因子和IgG的产生[32]。p38MAPK信号通路是LN炎症反应的一个关键途径[33-34]。本研究结果显示,p38MAPK的磷酸化水平在LN小鼠中的血清和肾脏组织中高表达。大量的研究已经表明,p38MAPK信号通路在促炎细胞因子合成前的转录和翻译过程中起着至关重要的作用,这使得p38MAPK信号通路的不同组成部分成为炎症性疾病和自身免疫性疾病潜在的治疗靶点[10,35]。自1993年发现以来,p38MAPK因其在广泛的细胞过程中的作用而备受关注,尤其是适应性免疫过程[2,6,36-39]。p38MAPK丰富地存在于T细胞和B细胞中[7-9,40];因此,p38MAPK信号通路必须在T细胞和B细胞的许多细胞过程和反应中发挥重要作用。有证据表明激活p38MAPK信号通路可以促进细胞凋亡[41]。另外,还有研究发现,LN患者外周血单个核细胞中磷酸化p38MAPK表达水平明显上调[32]。在本研究中,抑制和(或)敲低FKN的表达,则减少LN模型小鼠肾组织及Treg细胞中磷酸化p38MAPK的表达;此外,p38MAPK的激活剂可减少Foxp3及抗凋亡蛋白的表达;然而,p38MAPK的抑制剂则可逆转上述因子的表达。这说明,FKN可通过激活p38MAPK信号通路诱导Treg细胞的凋亡,减少Foxp3的表达。
综上所述,FKN可能通过激活p38MAPK信号通路参与Treg细胞的凋亡过程,从而参与LN的发生和发展。
