蛋白质错误折叠相关疾病的多肽药物研究进展*
2022-02-10吴思张红柯莎
吴 思 张 红 柯 莎
(1)中国科学院生物物理研究所,北京 100101;2)中国科学院大学生命科学学院,北京 100049)
蛋白质和多肽在特定条件下可能发生错误折叠,从可溶的天然态转化为不可溶的淀粉样聚集态。蛋白质的错误折叠和聚集与多种神经退行性疾病相关,包括常见的阿尔茨海默病(Alzheimer’s disease,AD)、帕金森氏病(Parkinson’s disease,PD)、亨廷顿病(Huntington’s disease)等[1‑2]。在这些疾病中,均有某种特定多肽或蛋白质从具有正常结构的可溶态转变为富含β折叠结构的淀粉样纤维沉积,从而导致细胞正常功能的丧失并引发细胞毒性。目前为止,已发现近40 种蛋白质或多肽形成的淀粉样纤维化沉积与退行性疾病相关,例如:AD 患者脑组织中的淀粉样斑块,主要由Aβ40 和Aβ42 多肽聚集形成,而患者神经细胞中发现的纤维缠结,则主要由Tau 蛋白聚集形成。PD 患者脑神经细胞中的Lewy小体,主要由α‑synuclein(α‑syn)聚集形成[1,3]。这些蛋白质在正常生理状态下具有不同的功能,例如:Aβ 被认为与中枢神经系统的保护相关,α‑syn 参与调节突触的功能,而Tau 作为微管结合蛋白,在促进微管组装和维持其稳定性方面发挥重要作用[4‑6]。这些蛋白质和多肽形成的淀粉样纤维具有相似的形态及结构特征,一般直径约5~20 nm,长度可达数微米,多肽链形成的β 折叠排列方向与纤维的轴向垂直。近年来,随着冷冻电镜技术的突破,使得包括Aβ、α‑syn、Tau 等多种退行性疾病相关的淀粉样纤维的高分辨结构得以解析[7‑15],为深入理解上述蛋白质的组装机制及其与不同疾病表型的关系,以及基于结构的药物设计提供了线索。
蛋白质的淀粉样纤维化是高度动态且复杂的过程,包含一系列可同时发生的微观反应步骤[16‑17],包括:蛋白质单体互相结合形成纤维核心(一级成核)、纤维的延伸生长、纤维的断裂、纤维表面催化单体生成新的纤维核心(二级成核)和纤维的末端重组等[18‑22],其中成核过程通常是限速步骤,核一旦形成即可作为模板使纤维化反应迅速推进。在上述复杂的动态过程中,存在多种不同结构和状态的中间体,从结构无序、大小不均一的蛋白寡聚体,到长度不同但结构有序的成熟纤维,多种聚集体形式可能共存,对于哪种组分是关键的致病因素仍然存在争论。目前为止的一些研究表明,淀粉样纤维化过程中的可溶的、可扩散的寡聚中间体可能是细胞毒性最强的组分[23‑25],它们可通过蛋白单体寡聚化生成,也可能由成熟纤维的断裂和解离产生。淀粉样蛋白寡聚体通过破坏细胞膜及内膜系统的完整性、导致膜的通透性改变以及细胞内钙离子失衡,引发线粒体功能异常及胞内大量活性氧(ROS)生成,进而引发细胞凋亡[26]。
由于淀粉样蛋白构象的无序性、聚集的动态性、过程的复杂性,以及致病组分尚不明晰,使药物研发工作充满困难和挑战[27]。在过去近30 年间,研究者们已针对蛋白质错误折叠和聚集相关疾病发展出多种不同的治疗策略,例如,干预蛋白质生成相关酶的活性以降低错误折叠蛋白质的生成水平;靶向错误折叠蛋白质或聚集体以增加其降解;稳定天然蛋白质结构和构象以降低错误折叠和聚集的倾向;增强和激活蛋白质质量控制体系以减少错误折叠和聚集;清除已形成的淀粉样纤维聚集体、阻断错误折叠寡聚体在细胞间传播等[28]。相关的试剂主要包括抗体、有机小分子和多肽等[29‑31],其中多肽药物因其高效性、可变和易修饰性、可降解性等诸多优势而备受关注[32‑35]。相比小分子药物,多肽具有更高的特异性和更低的毒性,而相比体积较大的抗体药物,多肽则具有较好的膜透过性、血脑屏障通过率以及较低的免疫原性,因而有望成为治疗蛋白质错误折叠和聚集相关疾病的候选药物。
由于多肽易被胃肠道的酶降解,口服利用率很低,因此一般采用注射方式。多肽类药物可分为天然多肽序列和多肽修饰物或类似物[29,36],主要以抑制淀粉样纤维化发生、减慢聚集体生长、降低淀粉样蛋白寡聚体及纤维的细胞毒性、促进高毒性寡聚体转化为低毒性组分为目的。本文将主要对AD相关的Aβ、Tau以及PD相关的α‑syn的多肽抑制剂相关研究进行总结,主要策略包括基于淀粉样纤维核心序列设计天然多肽,淀粉样纤维化核心序列多肽的修饰,以及随机筛选获得多肽。
1 基于淀粉样纤维化核心序列的天然多肽
通过对淀粉样蛋白成纤维核心的关键多肽序列进行筛选,从而获得有效的抑制剂前体,是一种常见的多肽抑制剂设计策略[37]。最早在Aβ淀粉样纤维化的研究中发现,Aβ纤维核心序列Aβ(16~20)五肽片段KLVFF,能够与全长Aβ40结合,破坏Aβ单体之间的有序组装,提示成纤维核心序列肽可对Aβ 的纤维化产生抑制作用[38]。以此为基础对KLVFF 多肽进行改造,在保持对成纤维核心序列识别能力的同时,增强多肽抑制剂的溶解性,加入破坏链间β 折叠的氨基酸及其他修饰[39‑46]。例如:通过加入脯氨酸残基破坏多肽形成β 折叠的倾向(如RDLPFFPVRID 和LPFFD)[40‑41];在多肽末端引入多个带电氨基酸残基破坏β 折叠(如KLVFFKKKK 和KLVFFEEEE),引起聚集形式改变,降低Aβ的细胞毒性[42]。其中,LPFFD首次在动物实验中显示,能有效减少Aβ 在大鼠脑组织中的沉积[41],因此被作为先导分子进行改造。如:N端和C端酰胺化的LPFFD能够降低AD转基因模型小鼠的脑损伤,并且比末端未修饰的LPFFD 具有更好的抗水解性,在血浆和脑脊液中可保持24 h不被降解[47];C 端酰胺化的LPYFD‑NH2等多肽,显示出对Aβ 纤维化聚集及神经细胞毒性的抑制作用,并且在小鼠实验中可缓解Aβ 聚集导致的记忆障碍[43‑44]。
采用纤维核心识别策略,研究者也设计了针对Tau 蛋白淀粉样聚集的多肽抑制剂。Tau 蛋白微管结合区(MTBR)是其淀粉样纤维的核心区域,位于MTBR 的两个六肽基序VQIVYK(PHF6,Tau(306~311))和VQIINK(PHF6*,Tau(275~280))是引发Tau 蛋白纤维化的关键序列[48‑49]。通过在VQIVYK序列的不同位置引入破坏β折叠结构的脯氨酸,发现其中Ac‑VPIVYK‑NH2和Ac‑VQPVYK‑NH2可抑制PHF6 聚集,还可解聚PHF6纤维并降低PHF6的神经细胞毒性[50]。但是后续研究发现,基于VQIVYK 设计的多肽只能抑制3R Tau(MTBR区含有3个重复序列的Tau蛋白)的聚集,并不能抑制全长Tau的聚集。因此,Eisenberg课题组[51]根据Tau 蛋白另一成纤维核心VQIINK所形成的“steric zipper”纤维结构设计了DVQMINKKRK(MINK)和 DVQWINKKRK(WINK),上述多肽在两倍过量的条件下对全长Tau 蛋白的淀粉样纤维化抑制率达50%,并能抑制纤维种子诱导细胞内Tau蛋白的聚集。进一步通过结构优化设计的W‑MINK多肽(DVWMINKKRK)显示出更强的阻断Tau 纤维种子传播能力,IC50约为1.1 μmol/L[51]。近期一项研究显示,Tau 蛋白MTBR 区的长片段Tau(258~360)在进行ΔK280以及I277P/I308P 突变后,在体外实验、N2a 细胞、以及线虫模型中均能够抑制Tau蛋白纤维化,且能够降低Tau 蛋白聚集的细胞毒性,改善Tau 蛋白聚集引起的线虫神经功能紊乱和运动障碍[52]。
α‑syn蛋白疏水性中心NAC片段是其淀粉样聚集的核心区域,其中α‑syn(68~76)和α‑syn(71~82)是形成淀粉样纤维的关键基序[53‑54]。Allsop课题组[55]从一系列α‑syn 的短肽片段中筛选出能与全长蛋白结合的α‑syn(68~72)片段GAVVT,并对其进行亲水性修饰,获得的RGGAVVTGR‑NH2以及RGAVVTGR‑NH2多肽可抑制α‑syn 淀粉样纤维化以及高分子量寡聚物的形成,进一步在该多肽C 端加入多个精氨酸以增加细胞膜的透过性(RGGACCTGRRRRRR‑NH2),可抑制M7‑A53T细胞中α‑syn‑A53T 聚集体形成和DNA 损伤,降低细胞毒性。Im 课题组[56]将NAC 区域的不同多肽片段中插入脯氨酸和带电荷氨基酸使之能破坏β折叠的形成,经过筛选发现含有第72 位丝氨酸(Thr)突变为脯氨酸(Pro)的6~10 肽均可抑制α‑syn 淀粉样聚集并能解聚成熟纤维,其中最短的6 肽PGVTAV经N端和C端酰胺化修饰,可在血浆中保持24 h 稳定,并抑制α‑syn 聚集引发的细胞毒性[57],有希望成为PD治疗的药物前体。近期的一项研究基于α‑syn(68~78)的纤维结构,将Thr72突变为Trp 并进行末端修饰,获得含 GAVVWGVTAV 序列的4 个多肽,其中 GAVVWGVTAVKKGRKKRRQRRRPQ(S62)与α‑syn纤维的解离常数可达0.5 μmol/L,在等摩尔条件下能抑制α‑syn淀粉样纤维化,显著降低α‑syn纤维种子诱导细胞内蛋白质聚集的能力[58]。
2 淀粉样纤维化核心序列多肽的修饰方式
成纤维核心序列本身往往具有较强的疏水性和聚集倾向,为提高多肽抑制剂的稳定性、水溶性和抗水解性,研究者对其进行了修饰和改造,主要包括下列方法:
2.1 氨基甲基化修饰
肽链骨架上的氨基(—NH—)甲基化能够阻断链间氢键网络的形成,因而有效抑制淀粉样纤维化[59‑61];同时,氨基甲基化多肽具有更高的水溶性和磷脂膜穿透能力[62]。交替氨基甲基化的多肽K(Me‑L)V(Me‑F)F(Me‑A)E‑NH2(Aβ(16~22m))能够结合Aβ40 纤维核心,抑制纤维生长延伸[60]。该甲基化多肽在水溶液中可达到40 g/L的浓度并以稳定的单体形式存在,与非甲基化的Aβ(16~22)相比,不易被蛋白酶降解[60]。在细胞实验中,Cruz 等[59]以KKLVFFA‑NH2为基础序列,对不同位置的氨基酸进行氨基甲基化,发现Phe 残基的氨基甲基化多肽(inL)可降低Aβ42的细胞毒性。类似地,在α‑syn 研究中,氨基甲基化 的α‑syn(68~78)(GAVVT(Me‑G)VTAVA)以及α‑syn(77~82)(VAQKT(Me‑V))多肽也均能有效抑制α‑syn 的纤维化[63‑64]。此外,与氨基甲基化类似的方法还包括将多肽链骨架上的酰胺键替换为酯键[65]。
2.2 D型多肽
天然氨基酸均为L型,L型多肽序列很容易被生物体内各种蛋白酶降解,使药效降低。而由D型氨基酸构成的多肽序列能够有效抵抗水解酶的作用,提高多肽抑制剂的代谢稳定性,降低免疫原性,增强通过血脑屏障的能力[66‑67]。研究发现,抑制Aβ 纤维化的多肽KLVFFA 的D 型对映异构体klvffa,比天然L 型多肽具有更高的抑制纤维化能力[68]。D 型N‑甲基化多肽(Me‑l)vffl‑NH2(PPI‑1019,Apan)进入了AD I期和II期临床试验,但随后被停止试验[69]。Doig 课题组[70]以KLVFF 为基础,对氨基酸的手性、氨基甲基化位置、乙酰化修饰、侧链修饰、肽链末端的酰胺化等不同条件进行系统的优化和筛选,发现C端酰胺化且单甲基化的D型多肽具有最好的效果;他们筛选获得高活性的 多 肽D‑[(chGly)‑(Tyr)‑(chGly)‑(chGly)‑(mLeu)]‑NH2(SEN304),该多肽能结合Aβ42并抑制其纤维化,促使毒性寡聚体转化为非毒性组分[71],SEN304 的衍生物SEN606 目前已处于临床前实验中[72]。Horsley 等[73]在研究中将D 型多肽klvff的末端氨基酸替换为D型的色氨酸,并在C端连接氨基丁酸(aminobutyric acid,Aib)作为破坏β折叠的基团,与天然的L型KLVFF相比,可更有效抑制Aβ42的成核和纤维延伸,显著降低Aβ42的细胞毒性。此外,研究还发现,纤维核心序列的反向D型多肽(retro‑inverso peptides)比天然正向多肽对于淀粉样蛋白核心序列有更强的亲和力,因此往往具有更好的抑制效果[74‑78]。Giralt 研究组[78]基于前期研究中获得的全L型氨基甲基化修饰多肽inL,获得其D 型对映异构体inD 以及反向D 型多肽inrD(a(Me‑f)fvlkk),比较结果显示inrD 比inL 和inD 具有更强的抑制Aβ42 细胞毒性的作用,且可抗胰蛋白酶等的水解。
在Tau 和α‑syn 多肽抑制剂研究中,基于纤维核心结构设计特异性结合的D型多肽也被作为一种重要策略。2011 年,Eisenberg 课题组[79]基于VQIVYK形成纤维的层间“steric zipper”结构,设计D型多肽tlkivw破坏β折叠堆积,阻断纤维延伸,该多肽与PHF6 纤维的解离常数约为2 μmol/L,可抑制PHF6以及Tau蛋白K12和K19截短体的聚集。Gazit课题组[80]从β‑syn序列中筛选与α‑syn结合的多肽片段,发现GVLYVGSKTR(β‑syn36)对α‑syn 纤维化和寡聚化具有最强的抑制作用,其D型异构体以及D型反向多肽在保持抑制能力的基础上,具有较强的抗水解性和细胞膜透过性,可降低果蝇PD模型中A53T突变型α‑syn的聚集,改善果蝇运动功能异常。上述研究结果表明,D型多肽的独特优势使之更有希望成为退行性疾病的备选药物。
2.3 多肽的环化
环化能够限制多肽的构象,使多肽抑制剂保持更高的稳定性。2003 年,Kapurniotu 等[81]使Aβ(1~28)中的Lys17 和Asp21 的侧链形成酰胺键,合成含有环结构的多肽cyclo(17,21)‑Aβ(1‑28),环化后的多肽自身不会聚集,且可抑制Aβ 的聚集并降低细胞毒性。环化的Aβ(16~20)多肽(cyclo‑KLVFF)及其D 型异构体对Aβ42 纤维化的抑制能力比线性KLVFF 提高了3 倍[82];进一步将此多肽第4 位Phe 残基的β 位连接苯环基团(cyclo‑KLVF(β‑Ph)F),可诱导Aβ42形成低毒性的、无法转化为纤维结构的“off‑pathway”寡聚体,从而有效抑制Aβ42发生纤维化[83]。在紫外光照射下,二氮环丙烯光活化基团修饰的cyclo‑KLVF(β‑Ph)F,可特异性地与Aβ42 的Tyr10 残基发生共价反应,降低Aβ42 的淀粉样聚集和细胞毒性[84]。
Nowick和Eisenberg研究组[85‑86]发展了一种称为“cyclic template”的环肽设计方法,为淀粉样纤维化抑制剂设计提供了新思路。环肽的一半由识别淀粉样纤维核心的片段组成,另一半含有被命名为“Hao”的三肽类似物作为骨架模板,这两部分自身能形成反向β 折叠结构,并阻断β 片层继续叠加(图1a)。这样的环肽结合在纤维种子末端,破坏氢键进一步形成,从而抑制纤维生长。Nowick等[85]采用上述策略,将Tau 蛋白PHF6 相关序列VQIVY 作为识别序列接入上述环肽骨架(图1b),该环肽分子对Ac‑PHF6‑NH2的纤维化有显著抑制作用。研究表明,环型结构对多肽的抑制效果起至关重要的作用,而环肽中关键残基的疏水性、手性等因素也与其抑制能力密切相关[85‑86]。Nowick等[87]改进了环肽骨架模板,将Aβ、α‑syn 等多种淀粉样蛋白的纤维核心作为识别序列,分别接入环肽分子;这些环肽分子在亚化学计量比水平即可对Aβ、α‑syn等的淀粉样纤维化起抑制作用,其中含有Aβ(16~22)即KLAFFAE 序列的环肽分子(图1c),能够显著降低Aβ40 和Aβ42 纤维化导致的细胞毒性。在近期的一项工作中,研究者将针对Aβ(16~21)和Aβ(37~42)所形成纤维结构的5 种多肽抑制剂片段接入环肽模板骨架,可显著增强其抑制Aβ42纤维化能力,在抑制剂与单体摩尔比为0.2的条件下,可使Aβ42 纤维化延滞期增加7~10 倍,抑制Aβ42形成寡聚体并降低其细胞毒性[88]。
除上述策略外,还可对多肽抑制剂进行其他化学修饰,或将其连接到纳米颗粒载体表面[89‑94],以增强多肽抑制剂的稳定性和抗水解性,提高其血脑屏障通过率,以达到更好的抑制效果。此外,将具有识别和抑制效应的多肽序列插入抗体中,可特异性结合淀粉样蛋白单体及其聚集物,为抗体药物的开发提供新思路[95‑97]。
3 随机筛选
除了基于淀粉样纤维核心的序列和结构进行理性设计得到多肽抑制剂之外,随机筛选也是发现有效多肽抑制剂的方法之一[67,98‑105]。镜像噬菌体展示(mirror phage display)是常用的多肽筛选技术[106]。Willbold 课题组[100,107‑108]采用镜像噬菌体展示技术,从12肽随机序列中筛选对Aβ42纤维具有高亲和力的序列,筛选出D 型多肽qshyrhispaqv(D1)和rprtrlhthrnr(D3),二者均可特异性识别APP/PS‑1转基因小鼠模型脑组织中的Aβ42淀粉样斑块,降低Aβ 聚集的细胞毒性。D1 对Aβ 纤维具有更高的亲和力,能够增加Aβ42 聚集体的数量,但显著减低聚集体的大小;D3则对Aβ寡聚体亲和力更高,可改变Aβ42 聚集体形态,使之形成无毒性的无定形沉淀[99‑100,109]。其中,D3在临床前实验中具有减少转基因小鼠脑组织中Aβ 淀粉样斑块形成、降低炎症反应的效果;同时D3 具有良好的抗水解稳定性和血脑屏障通过率,口服利用率可达58%[110‑113],因此D3 及其衍生物有望成为治疗AD的候选药物[114‑115]。此外,在Tau 蛋白相关的研究中,Funke 课题组[67]利用镜像噬菌体展示技术,从109条12 肽中筛选出6 个与PHF6 纤维结合的多肽序列,其中D 型多肽ttslqmrlyypp(TD28)和ppyylrmqlstt(TD28rev)具有较强的抑制Tau 蛋白纤维化作用以及良好的细胞膜透过性。近期,该课题组又以PHF6*形成的纤维作为靶标,通过镜像噬菌体展示筛选获得D 型多肽dplkarhtsvwy(MMD3)及其反向序列(MMD3rev),它们能够抑制PHF6*及全长Tau蛋白的纤维化,使之形成无定形沉淀[102]。将靶向PHF6 和PHF6*的序列整合在同一分子上,也可能产生更有效的针对Tau蛋白聚集的药物。
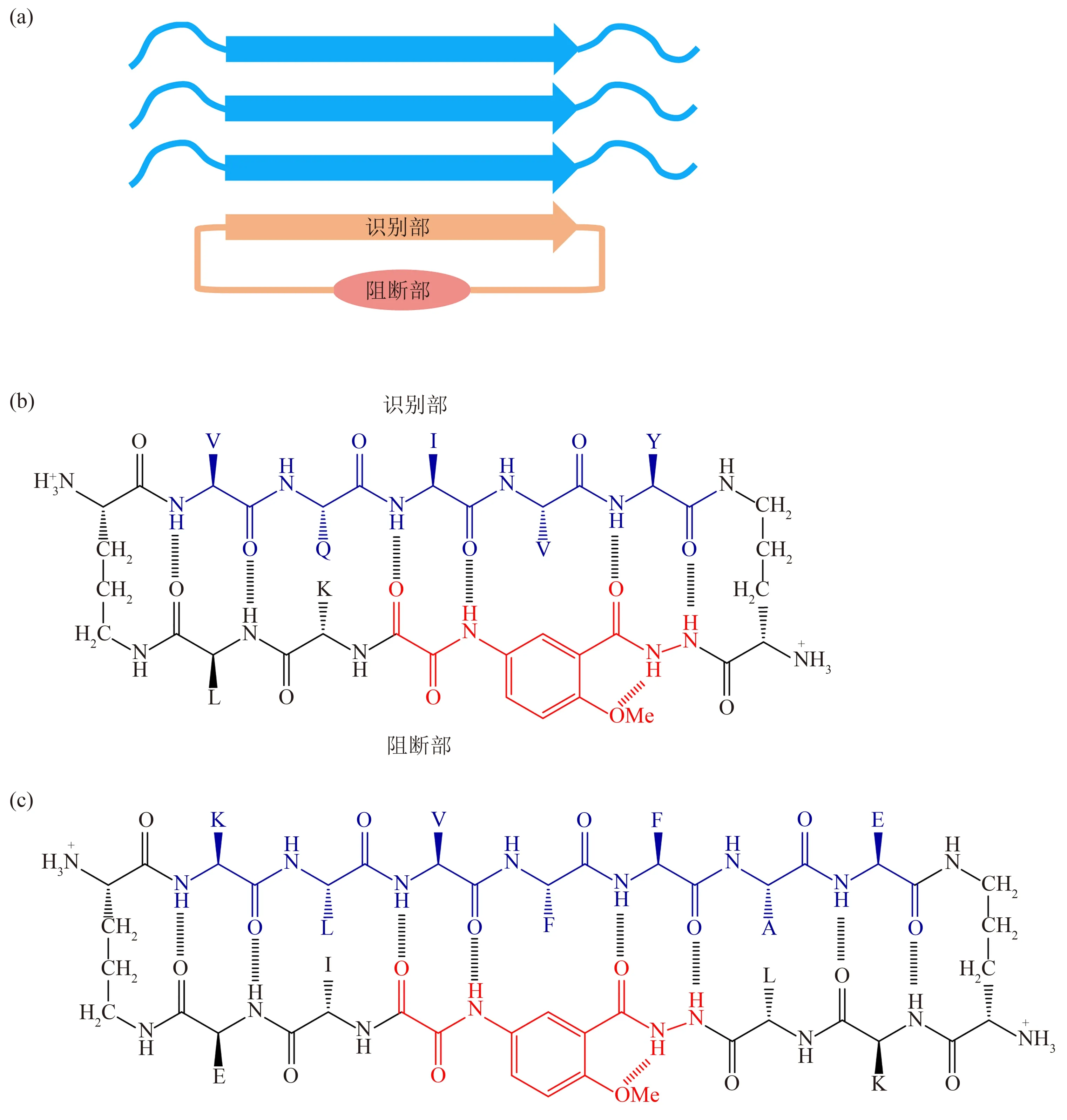
Fig.1 Design of cyclic-peptide inhibitors of amyloid formation图1 抑制淀粉样纤维化的环肽分子设计
4 其他新思路
设计靶向毒性相对较高的淀粉样蛋白寡聚体的抑制剂,是近年多肽药物研究的新思路。Daggett等[116‑117]研究发现,Aβ 等多种不同的淀粉样蛋白在聚集早期可形成具有非典型二级结构“α‑sheet”的毒性寡聚体。研究者据此设计出能够形成α‑sheet 的多肽片段,如AP5、AP90、AP407 和AP421[116],它们对单体和纤维的亲和力较低,但能以纳摩尔级亲和力特异性结合Aβ 寡聚体,可抑制Aβ的聚集和引发的神经细胞毒性,并且抑制Aβ聚集导致的转基因线虫麻痹症,促进AD小鼠模型中毒性Aβ 寡聚体的清除,有望成为药物和诊断试剂的前体[116]。在近期的一项关于α‑syn 蛋白的多肽抑制剂研究中,Santos等[118]发现,采用具有正电性和两亲性的α螺旋型的天然多肽PSMα3,虽不与天然态α‑syn 单体结合,但能够以纳摩尔级亲和力特异性结合α‑syn 蛋白的寡聚体和纤维聚集体,并在亚化学计量比的条件下抑制α‑syn 的聚集,且消除寡聚体对神经细胞的毒性。通过分析PSMα3与α‑syn 寡聚体及纤维相互作用的界面特征,设计出多个具有类似效果的多肽,并通过筛选发现人源抗菌肽LL‑37 也符合上述特征,它们均能与α‑syn的毒性寡聚体相互作用,并显著降低细胞毒性。有趣的是,LL‑37 多肽也被发现能够抑制Aβ 和IAPP的淀粉样纤维化[119‑120]。相比外源多肽抑制剂,内源性多肽具有更好的生物相容性,更有潜力成为疾病治疗的药物。
越来越多的证据显示,淀粉样蛋白所形成的寡聚体可能比成熟纤维具有更高的毒性,加速寡聚体向成熟纤维的转化也被作为治疗蛋白质错误折叠疾病的潜在策略[121‑124]。本课题组[125]在近期关于酵母prion蛋白Ure2淀粉样纤维化机制的研究中,从Ure2 成纤维核心区域筛选出LQVNIGNR 多肽,该多肽能够组装成泡状结构,加速Ure2、Tau 以及α‑syn 的纤维化过程。通过单分子荧光实验和全局动力学建模分析,揭示LQVNIGNR 多肽可促进高毒性寡聚体向低毒性纤维转化,从而减少寡聚体累积并降低细胞毒性。通过催化机制加速毒性组分转化为无毒性或低毒性组分,相比通过结合单体及中间体而抑制纤维生长所需的抑制剂剂量更低,使毒性中间体组分存在的时间更短(图2),为神经退行性疾病的防治提供了新思路。

Fig.2 Difference between the working mechanisms of catalysts and inhibitors of amyloid formation图2 催化剂与抑制剂作用机制的差异
5 展望
多肽是较早开始发展的针对蛋白质错误折叠相关疾病的药物前体,截至目前已开展了大量的研究工作。因多肽具有高特异性、低毒性、低免疫原性、易修饰性,以及生物兼容性等优势,它们有望成为治疗退行性疾病的先导分子和备选药物[29,35]。本文总结了过去20 多年中,针对Aβ、Tau 以及α‑syn淀粉样纤维化的多肽抑制剂研发的主要策略,以及对基于纤维核心的合理设计和随机筛选等方式获得的多肽抑制剂进行修饰和优化的方法(表1),这些研究结果为进一步开发有效药物奠定了良好的基础。然而,目前多数研究仅停留在体外实验及细胞实验水平,仅有少数多肽进入临床前研究,尚未成为临床确证有效的药物[126‑127]。尽管退行性疾病的致病机制尚未完全清楚,仍然存在争论[128],但大量的研究证据表明,蛋白质淀粉样聚集的过程与退行性疾病的发病密切相关[28]。鉴定蛋白质聚集过程中不同组分的细胞毒性及其时空因果关系,深入揭示蛋白质聚集体的结构及其与毒性的关联,对于发展有效的治疗策略起关键作用。在未来的研究中,包括干扰淀粉样纤维化过程在内的多种治疗策略的联合应用将有望为退行性疾病治疗带来新希望。同时,基于淀粉样纤维化过程开发疾病检测和诊断的生物标志物,对于退行性疾病的早期诊断以及评判临床治疗效果也有十分重要的意义。

Table 1 Peptide inhibitors targeting Aβ,Tau and α-syn fibril formation表1 针对Aβ、Tau、α-syn纤维化的多肽抑制剂
猜你喜欢
杂志排行

生物化学与生物物理进展的其它文章
- Protein Aggregation and Phase Separation in TDP-43 Associated Neurodegenerative Diseases*
- 细菌的信号转导系统及其在耐药中的作用*
- CRBGP Inhibited The Activity of Glioma U251 Cells Through Suppressing FAK-AKT Pathway and The Secretion of Interleukin-6*
- Bradykinin Upregulated The Expression of Cyclooxygenase-2 in The Submucosal Plexus of Enteric Nervous System of Guinea Pig*
- 阿尔茨海默病体外诊断纳米技术*
- α-Synuclein as a Diagnostic Marker and Therapeutic Target for Parkinson Disease*