Lack of transcriptional coordination between mitochondrial and nuclear oxidative phosphorylation genes in the presence of two divergent mitochondrial genomes
2022-01-27RanXuMariangelaIannelloJustinHavirdLilianaMilaniFabrizioGhiselli
Ran Xu, Mariangela Iannello, Justin C.Havird, Liliana Milani, Fabrizio Ghiselli
1 Department of Biological, Geological and Environmental Sciences, University of Bologna, Bologna, BO 40126, Ιtaly
2 Department of Ιntegrative Biology, The University of Texas at Austin, Austin, TX 78712, USA
ABSTRACT In most eukaryotes, oxidative phosphorylation(OXPHOS) is the main energy production process and it involves both mitochondrial and nuclear genomes.The close interaction between the two genomes is critical for the coordinated function of the OXPHOS process.Some bivalves show doubly uniparental inheritance (DUI) of mitochondria, where two highly divergent mitochondrial genomes, one inherited through eggs (F-type) and the other through sperm (M-type), coexist in the same individual.However, it remains a puzzle how nuclear OXPHOS genes coordinate with two divergent mitochondrial genomes in DUI species.In this study,we compared transcription, polymorphism, and synonymous codon usage in the mitochondrial and nuclear OXPHOS genes of the DUI species Ruditapes philippinarum using sex- and tissuespecific transcriptomes.Mitochondrial and nuclear OXPHOS genes showed different transcription profiles.Strong co-transcription signal was observed within mitochondrial (separate for F- and M-type) and within nuclear OXPHOS genes but the signal was weak or absent between mitochondrial and nuclear OXPHOS genes, suggesting that the coordination between mitochondrial and nuclear OXPHOS subunits is not achieved transcriptionally.McDonald-Kreitman and frequency-spectrum based tests indicated that M-type OXPHOS genes deviated significantly from neutrality, and that F-type and M-type OXPHOS genes undergo different selection patterns.Codon usage analysis revealed that mutation bias and translational selection were the major factors affecting the codon usage bias in different OXPHOS genes, nevertheless, translational selection in mitochondrial OXPHOS genes appears to be less efficient than nuclear OXPHOS genes.Therefore, we speculate that the coordination between OXPHOS genes may involve posttranscriptional/translational regulation.
Keywords: Oxidative phosphorylation; Doubly uniparental inheritance; Co-transcription; Polymor-phism; Codon usage bias; Translational selection
lNTRODUCTlON
In most animals, mitochondria are maternally inherited and one consequence of this kind of inheritance is a limitation of the genetic variance in the mitochondrial population within an individual (Lane, 2012).Until recently, it was assumed that mitochondrial DNA (mtDNA) is present in a state of homoplasmy, namely that identical copies of the mtDNA are found within an individual.The presence of different genetic variants, termed heteroplasmy, was thought to be mainly associated with unfavorable conditions such as ageing and disease (e.g., James White et al., 2008; Lane, 2012; Stewart& Chinnery, 2015).More recently, high-throughput sequencing revealed that mtDNA heteroplasmy (at least at low levels) is much more common than previously thought (Barrett et al.,2019; Dowling, 2014; Zhang et al., 2018).Heteroplasmy is a central issue in mitochondrial biology because genetic variation can lead to within-individual selection which can negatively affect coordination with nuclear-encoded mitochondrial components (Lane, 2011).
In Metazoa, the primary function of mitochondria is energy production through the process named oxidative phosphorylation (OXPHOS), which involves the tight interaction between mitochondrial (mt) and nuclear (nu)encoded subunits.Therefore, to ensure the correct and efficient synthesis and assembly of the OXPHOS system,proper coordination between mt and nuOXPHOS genes is required.In mice and humans, van Waveren & Moraes (2008)reported a shared transcriptional control mechanism of nuOXPHOS genes and strongly correlated transcriptional signal within the same complex of nuOXPHOS genes.Barshad et al.(2018) investigated the OXPHOS transcription regulatory landscape across multiple tissues in humans and found a strong co-regulation signal between mt and nuOXPHOS genes across tissues.These findings make intuitive sense as gene products that must cofunction may be transcriptionally co-regulated.However, Couvillion et al.(2016) showed that transcription levels in mt and nuOXPHOS genes were not concordant during mitochondrial biogenesis in yeast, while translational responses in both mt and nuOXPHOS genes were rapid and synchronously regulated,indicating the coordination between mt and nuOXPHOS genes is at the translation, not transcriptional level.A recent study in human cells showed that the average synthesis of mt and nuOXPHOS subunits for each complex was also highly correlated, although coordinated cytosolic and mitochondrial translation may require a nu-encoded mt protein—leucine rich pentatricopeptide repeat containing protein (LRPPRC)—to maintain cellular proteostasis (Soto et al., 2021).
Transcriptional coordination between mt and nuOXPHOS genes could be particularly complex in some bivalve species.Some bivalve species exhibit an evolutionarily stable exception to the strictly maternal mtDNA inheritance (SMI), a condition referred to as doubly uniparental inheritance (DUI;see Zouros, 2013 for a review).In DUI species, two mitochondrial lineages (F-type and M-type) are present: the F-type is transmitted through eggs while the M-type is transmitted through sperm.Because of the strict segregation between F and M lineages, F- and M-type mtDNA can accumulate an impressive genetic divergence (10%–43% at the nucleotide level, up to 53% at the amino acid level).Gametes are homoplasmic for the respective sex-linked lineage, while the distribution of F- and M-type in adult tissues is variable according to sex, tissue, and species.Generally,heteroplasmic females are less common (and if heteroplasmy is present it is usually at low levels), whereas males are always heteroplasmic.According to previous studies, the M-type mtDNA in the DUI species is predominant in male gonads and present in variable number (and can also be absent) in male somatic tissues, being generally absent (or rare) in female samples.F-type mtDNA is present in all the somatic tissues of both females and males (Ghiselli et al., 2019,2021a).Moreover, the transcription of M-type mtDNA was also detected in male somatic tissues, but the frequency and the percentage of its presence seem to vary across different species (e.g., Breton et al., 2017; Dalziel & Stewart, 2002;Ghiselli et al., 2011; Milani et al., 2014; Mioduchowska et al.,2016; Obata et al., 2011).For example, the M-type mtRNA was detected in 60% and 89.5% of male somatic samples in Utterbackia peninsularis and Venustaconcha ellipsiformis,respectively (Breton et al., 2017).However, sex- and tissuespecific transcriptomic resources are lacking for DUI species,making many inferences on the abundance and expression of F-type, M-type, and related nuclear genes difficult.
While mito-nuclear coregulation is likely important, it is therefore unclear when and where selection has acted on coordination across different eukaryotes.Sequence coevolution between mt and nuOXPHOS genes provides another way to coordinate OXPHOS across the genomes.Mito-nuclear coevolution implies that sequence evolution within one genome could exert selection on the other genome for complementary changes (Hill, 2020; Hill et al., 2019; Rand et al., 2004), and mito-nuclear coevolution has been observed across a wide range of eukaryotic lineages (Barreto et al.,2018; Barreto & Burton, 2013; Havird et al., 2017; Havird &Sloan, 2016; Yan et al., 2019), including bivalves (Piccinini et al., 2021, which included 4 DUI species).In DUI species, two highly divergent mt genomes have to cofunction with the same nuclear background, which may be challenging for mitonuclear coevolution.Previously studies indicated that F- and M-type genomes might have evolved separately multiple times(Gusman et al., 2016; Zouros, 2013), and two types of mt genomes might be under different selective pressure.It has been reported that M-type mt genomes show higher nonsynonymous to synonymous substitution rates (dN/dS)than F-type, and several studies proposed that the M-type mt genome might be under relaxed selection (Hoeh et al., 1996,1997, 2002; Liu et al., 1996; Ort & Pogson, 2007; Śmietanka et al., 2009, 2013; Soroka & Burzyński, 2010; Stewart et al.,1995, 1996; Zbawicka et al., 2010; Zouros, 2013).However,other studies have hypothesized that M-type may have undergone adaptive evolution optimizing sperm/male gonad functions (Bettinazzi et al., 2019, 2020; Burt & Trivers, 2006;Ghiselli et al., 2013, 2021a, 2021b; Iannello et al., 2019;Skibinski et al., 1994, 2017).Therefore, studying selection on different OXPHOS genes would be critical to understandinghow two divergent mt genomes evolve with the same nuOXPHOS genes (Iannello et al., 2019; Maeda et al., 2021;Piccinini et al., 2021).
An assumption of dN/dS metrics is that variations at synonymous sites are neutral.However, recent studies have shown selection on synonymous sites in the form of preferential codon usage, without changing the protein sequence (reviewed in Gingold & Pilpel, 2011; Plotkin &Kudla, 2011).Natural selection acting on synonymous codons to increase protein synthesis speed and accuracy is known as translational selection.Translational selection combined with mutational bias can create synonymous codon usage bias(CUB), in which codons are used in different frequencies in the coding regions across the genome (Gingold & Pilpel,2011; Hershberg & Petrov, 2009; Plotkin & Kudla, 2011).CUB can influence various cellular processes, including gene expression (Jeacock et al., 2018), protein folding and function(Yu et al., 2015; Zhou et al., 2009), and exon splicing(Parmley & Hurst, 2007), therefore it can play an important role in genome evolution.In addition, it has been shown that translational selection is pervasive and detectable in a wide range of vertebrates (de Oliveira et al., 2021; Doherty &McInerney, 2013; Machado et al., 2020).Translational selection has also been detected in mitochondria in a wide range of species (Jia & Higgs, 2008; Sun et al., 2008; Wang et al., 2011; Wei et al., 2014).Furthermore, several studies reported that the rate of mRNA translation into protein(translational efficiency) in mt genes is lower than nuclear genes (Adrion et al., 2016; Havird & Sloan, 2016; Pett &Lavrov, 2015; Sloan et al., 2013), leading to the hypothesis that differences in translational selection for efficiency between mt and nu genes might be associated with the different evolution rates in mt and nuOXPHOS genes.
DUI species, with the stable and natural occurrence of two very divergent mitochondrial genomes in the same individual,represent an interesting evolutionary puzzle, and provide a unique model to study heteroplasmy and mito-nuclear interactions.Taking advantage of RNA-Seq data on three different tissues of 15 females and 15 males, we compared transcription, polymorphism, divergence, and codon usage in mt and nuOXPHOS genes in the DUI species Ruditapes philippinarum (the Manila clam).We observed lack of cotranscriptional coordination among F-type, M-type and nuOXPHOS genes.Furthermore, three genomes were constrained by different selection patterns, occurring at both synonymous and nonsynonymous substitutions.In particular,transcriptional selection shapes codon bias differently in mt and nuOXPHOS genes.Considering our results, we predict that mito-nuclear coordination does not occur at transcriptional level, but it is achieved by post-trascriptional/translational mechanisms in DUI species.To our knowledge, this is the first study analyzing both transcriptional regulation and sequence evolution to investigate the coordination of OXPHOS genes in mollusks.
MATERlALS AND METHODS
Dataset and reference transcriptome
Raw reads of Ruditapes philippinarum were downloaded from NCBI (BioProject PRJNA672267).All the clams were collected during the spawning season (end of July) from the same population in the Northern Adriatic Sea (Italy), in the river Po delta region (Sacca di Goro, approximate GPS coordinates:N44°50 ′06 ″, E12°17 ′55 ″).By visual inspection at optical microscope, gonads contained either eggs or sperm in late developmental stages, and clams were sexed concordantly.Adductor muscle, mantle, and gonad from 15 males and 15 females (with the exception of a missing female mantle; 89 samples in total) were sequenced using Illumina HiSeq 2500 with insert size of 500 bp to generate 150 bp paired-end reads.Detailed information about RNA extraction, library preparation, sequencing and de novo transcriptome assembly can be found in Maeda et al.(2021).The de novo reference transcriptome assembly for R.philippinarum is available on the Transcriptome Shotgun Assembly Sequence Database(TSA) of NCBI (accession No.GIVW00000000).
Transcriptome analysis
We used CD-HIT-EST (Fu et al., 2012) to reduce transcriptome redundancy (the presence of multiple transcripts belonging to the same gene), with a similarity threshold of 0.9.To retrieve F- and M-type mt genes from the transcriptome,we downloaded the complete F- and M-type mt genomes of R.philippinarum from NCBI (Accession Nos.: AB065374,AB065375).Considering some inaccuracies in the NCBI original annotations, we reannotated the mt genomes, by using BLAST (Camacho et al., 2009) against the nonredundant protein database (nr) to confirm protein coding regions, and by manually curating start and stop codons(Supplementary File 1).Then all reads were mapped to the transcriptome (without mt transcripts) and reference mt genes.The filtered reads were mapped to the transcriptome using bowtie2 (Langmead & Salzberg, 2012) with the default settings and only reads with mapping quality >10 were included in the following analyses.SAMtools (Li et al., 2009)was used to retrieve reads that were properly paired and uniquely mapped.Samples having <1 thousand reads mapped to mt protein coding genes and <1 million total mapped reads, were excluded from the analysis.
Transcriptome annotation
Nuclear OXPHOS transcripts were retrieved from Maeda et al.(2021) (BioProject PRJNA672267).To get coding sequences from nuOXPHOS transcripts, we ran TransDecoder(https://github.com/TransDecoder/TransDecoder/wiki) using homology searches against nr and Pfam databases with the minimum length of the open reading frame of 150 bp and only the longest ORF for each OXPHOS gene was kept.
We additionally performed the annotation of the whole R.philippinarum transcriptome as follows.First, contaminations from non-metazoans were filtered out by using a BLASTX(Altschul et al., 1997) search (with default parameters and adding information about taxon id) against the nr database of NCBI.We therefore extracted the full taxonomic lineage for each BLAST hit and we kept only transcripts having a best BLAST hit against Metazoa.To predict open reading frames(ORF) in the transcriptome, we used findorf (Krasileva et al.,2013); the prediction was performed using both a BLASTXsearch against the nr database and an HMMER (Mistry et al.,2013) search against the Pfam database (Finn et al., 2016).Annotation of predicted proteins was performed by using both a BLASTP search against the Swiss-Prot database and an HMMER search against the Pfam database.We used Argot2(Falda et al., 2012) to obtain Gene Ontology (GO) terms from BLASTP and HMMER outputs.
Transcription and co-transcriptional analysis
We used the gene length-corrected TMM (GeTmm)normalization method to allow both intra- and inter-sample comparisons (Smid et al., 2018).Before normalization,transcripts with low number of counts were filtered out using the NOIseq R package (Tarazona et al., 2015), with the following parameters: CPM=1, cv.cutoff=300.Genes that failed to pass this threshold were defined as lowly transcribed genes.It is worth mentioning that F-type and M-type genomes both contain a lineage-specific ORF (FORF and MORF),which might play functional roles in DUI species (Breton et al.,2011; Milani et al., 2013a; Minoiu et al., 2016).Therefore,although we still do not know if they belong to any complex subunits, we included them among the mtOXPHOS genes if not specified in the context.The transcription for mt and nuOXPHOS genes were plotted for each tissue, and Wilcoxon rank-sum test was used to compare pairwise transcriptional differences between mt and nuOXPHOS genes in each tissue.Kruskal-Wallis rank-sum test followed by a Dunn‘s test with Bonferroni correction was used to compare transcriptional differences across tissues.
To retrieve the general correlation trend of transcription across tissues, we calculated Spearman‘s rank-sum correlation with FDR correction across all samples using psych R package (http://CRAN.R-project.org/package=psych).The same process was also performed separately for each tissue to obtain the tissue-specific correlation trend.The correlation was considered significant with adjusted P<0.05.To test if the correlation strength between mt and nuOXPHOS genes was higher than genes involved in the different biochemical activities, we compared the differences in Spearman‘s correlation coefficients (rho) between OXPHOS genes and a set of randomly selected same number of nuclear genes (56 genes) using Wilcoxon rank-sum test with Bonferroni correction.To test the hypothesis that genes within the same complex (intracomplex) presented a stronger correlation than the genes between different complexes(intercomplex), we compared the intracomplex correlation to intercomplex correlation using Wilcoxon rank-sum test with Bonferroni correction.
To uncover genes co-transcribed with OXPHOS genes, we retrieved the nuclear genes that were significant highly(rho>0.6) correlated with mtOXPHOS genes, and with nuOXPHOS genes.Functional enrichment for the nuclear genes that were co-transcribed with the OXPHOS genes was performed in topGO v2.34.0 (Alexa & Rahnenführer, 2018),using the classic Fisher‘s test, with a nodeSize of 5 and a P-value cutoff of 0.01.
SNP calling and McDonald-Kreitman test
The F- and M-type mt genomes were used as references for SNP calling on mtOXPHOS genes.For the nuOXPHOS genes, SNPs were called based on the de novo assembled transcriptome.We used Freebayes v1.2.0 (Garrison & Marth,2012) to call the SNPs from all the samples simultaneously.VCFtools (Danecek et al., 2011) was run to calculate the rate of missing SNPs and to filter the SNPs for each sample using the following parameters: --minGQ 20 --minQ 30 --minDP 30.Finally, the number of SNPs in each gene was normalized by the gene length and the total number of SNPs in the sample to enable comparison across different genes.Considering the uneven coverage and the different rates of missing SNPs across sexes and tissues, allele frequency and PCA classification were performed with a genotype likelihood approach implemented in ANGSD (Korneliussen et al., 2014),which is particularly suited for low and medium depth data.The Kolmogorov-Smirnov test with Bonferroni correction was used to assess if there is difference in the distribution of allele frequencies between different set of genes.SnpEFF(Cingolani et al., 2012) was used to predict the effects of SNPs on mt and nuOXPHOS genes.Samples with the rate of missing data >40% were filtered out for the SNP effect prediction.Statistical differences between the proportion of synonymous and nonsynonymous SNPs in each component of OXPHOS (F-type, M-type and nuOXPHOS) genes were tested by Wilcoxon rank-sum test with Bonferroni correction.
We performed McDonald-Kreitman (MK) tests (McDonald &Kreitman, 1991) and frequency-based tests—Tajima‘s D(Tajima, 1989), Fu & Li‘s D, and Fu & Li‘s F (Fu & Li,1993)—on mt and nuOXPHOS genes and randomly selected nuclear protein-coding genes (30 genes), using DnaSP v6.12(Rozas et al., 2017).For mtOXPHOS genes, the MK test was performed between F-type and M-type OXPHOS, and also separately for F-type and M-type using the closely related species Ruditapes decussatus (SMI) as an outgroup.For nuOXPHOS and randomly selected genes, the MK test was performed with R.decussatus as an outgroup.The OXPHOS orthologues in R.decussatus were extracted from Iannello et al.(2019) and Piccinini et al.(2021), while the random orthologues were retrieved with OrthoFinder2 (Emms & Kelly,2019).Among the nuOXPHOS genes identified above, 32 nuOXPHOS orthologues were retrieved for the MK analysis.VCFtools (Danecek et al., 2011) and SeqKit (Shen et al.,2016) were used to retrieve consensus nucleotide sequences and amino acid sequences, respectively.Clustal Omega(Sievers & Higgins, 2018) was used for multiple sequence alignment and PAL2NAL (Suyama et al., 2006) was used to retrieve the homologous nucleotide region.
Codon usage analysis
The codon frequencies of OXPHOS genes were calculated using the EMBOSS cusp tool (http://emboss.bioinformatics.nl/cgi-bin/emboss/cusp).The genetic codes 1 and 5(https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi)were used for the nuclear genes and mitochondrial genes,respectively.The GC composition (at first and second codons position: GC12, at the third codon position: GC3), relative synonymous codon usage (RSCU), the effective number of codons (ENC), and codon adaptation index (CAI) were calculated using CAIcal (Puigbò et al., 2008).RSCU is definedas the ratio of the observed frequency of codons to the expected frequency given that all the synonymous codons for the same amino acid are used equally (Sharp & Li, 1987).The ENC quantifies the extent to which the codon usage in a gene or genome departs from the equal usage and it ranges from 20 (if only one codon is used for each amino acid) to 61 (if all codons are used equally) (Wright, 1990).CAI is another commonly used statistic which requires a set of highly expressed genes as reference and it presumes translational selection in highly expressed genes, therefore it can assess the extent to which selection has driven the pattern of codon usage (Sharp & Li, 1987).For the mitochondrial genes, the reference database for CAI estimation is available.Considering M-type OXPHOS genes were present mainly and predominantly in male gonads, we therefore only used the average transcription levels in male gonads for M-type OXPHOS genes and in female gonads for F-type OXPHOS genes to calculate the correlation between CAI and the transcription levels.For nuclear genes, no reference database was available, so 30 highly transcribed (average transcription in gonads) nu-encoded genes: top 15 transcribed nuOXPHOS genes and top 15 transcribed nu-encoded genes (ribosomalrelated genes were excluded to avoid bias) were selected to build the reference database (Supplementary File 2).We also calculated ENC and CAI for the whole transcriptome, and Kruskal-Wallis rank-sum test followed by a Dunn‘s test with Bonferroni correction was performed to assess if ENC and CAI presented differences between OXPHOS genes and nuclear genes.The ENC-GC3 relationship (Nc-plot, Wright,1990) and neutrality test between GC12 and GC3 (Sueoka,1999) were performed to assess the factors influencing the CUB in OXPHOS genes.Spearman‘s rank-sum test was used to assess the relationship between ENC and CAI.The comparison between ENC and CAI was also used to demonstrate the relationship between mutation and natural selection on codon usage bias (Behura & Severson, 2012).The correspondence analysis (COA) based on both codon counts and RSCU was performed in R FactoMineR package to detect the factors affecting CUB.We also performed Chisquare tests for context-dependent mutations (the rate of mutation from any one base to any other is influenced by the neighboring bases) in each set of OXPHOS genes according to the procedures described in Jia and Higgs (2008).The mutation equilibrium was calculated according to Lynch(2007).Briefly, we inferred the minor allele according to the allele frequency and treated the minor alleles as the new mutations (Hildebrand et al., 2010).Sites with more than two alleles or with two alleles at an equal frequency were discarded.Then the expected mutation GC equilibrium was calculated as the following formula:

where m=v/u, u is the mutation rate of A/T to G/C and v is the mutation rate of G/C to A/T (Johri et al., 2019; Lynch, 2007).Consequently, m close to 1 indicates little mutation bias, while GCeq close to the percentage of GC3 means that codon usage bias is majorly determined by mutation bias (Johri et al.,2019; Lynch, 2007).
RESULTS
Different transcription patterns of mitochondrial and nuclear OXPHOS genes in different tissues
Five samples were filtered out due to a low number of reads or contamination (f_67_G, f_67_M, m_70_A, m_70_G, m_70_M;Supplementary Table S1).A total number of 84 samples was used for the following analysis.The percentage of reads mapped to the F- and M-type mtOXPHOS genes were reported in Supplementary Figure S1: F-type was predominant(~100% in females; >95% in male somatic tissues) in all the tissues except male gonads, while M-type accounted for more than 90% of reads in male gonads.Small traces of M-type reads were also detected in male adductor muscles (average:0.92%) and male mantles (average: 0.58%) with five samples presenting more than 1% of M-type reads in male somatic tissues (Supplementary Figure S1 and Table S2).The median depth for each gene was calculated for all the samples(Supplementary Table S2).No read was retrieved for the ATP8 gene, possibly due to its short length (120 bp in F-type and 84 bp in M-type).
After filtering lowly transcribed genes, 27 mitochondrial protein-coding genes (14 from F-type and 13 from M-type), 56(out of 67) nuOXPHOS genes, and 20 214 nuclear encoded genes were kept.The normalized counts for the OXPHOS genes are reported in Supplementary Table S3.To assess transcriptional differences of OXPHOS genes across different components (F-type, M-type and nuOXPHOS), a PCA analysis was performed based on the transcription level of all OXPHOS genes (Figure 1A–C), only nuOXPHOS genes(Supplementary Figure S2A) and only mtOXPHOS genes(Supplementary Figure S2B–C).We found that F-type, M-type, and nuOXPHOS formed three distinct clusters taking account of transcription of all the OXPHOS genes across tissues (Figure 1A).Notably, M-type OXPHOS genes were clustered remarkably apart from the F-type and nuOXPHOS genes, in line with our expectations because the transcription of M-type in somatic tissues is rare or absent (Supplementary Tables S2, S3).However, transcriptional profiles of M-type in male gonads can also contribute to this departure.To test this,we conducted the PCA analysis for all the OXPHOS genes only in male gonads.M-type presented an extremely wide distribution despite the overlap between different OXPHOS genes in male gonads (Figure 1B), indicating that transcriptional profiles of M-type in male gonads also contributes partially to the departure of M-type in Figure 1A.Moreover, the OXPHOS genes in different tissues also showed different patterns, with male gonads departing from the other tissues and adductors showing relatively wider variation (Figure 1C).To further investigate transcriptional differences across tissues (Figure 1C), we focused on the nu and mtOXPHOS genes separately.Interestingly, we found that the distribution of nuOXPHOS genes showed an overlap in different tissues despite the wider variation in adductor muscles (Supplementary Figure S2A).By contrast, if we consider all the mtOXPHOS genes together, we found that male gonads were clearly apart from the other tissues(Supplementary Figure S2B).However, if we only look at F-type OXPHOS genes, the male gonads did not deviate fromthe other tissues (Supplementary Figure S2C).Thus, it seems that the deviation of male gonads in Figure 1C and Supplementary Figure S2 was due to the presence of M-type OXPHOS genes.

Figure 1 The PCA plot based on the transcription of OXPHOS genes
Figure 2 compares the transcription level of mt and nuOXPHOS genes in different tissues.Because we do not know whether the two lineage-specific ORFs (FORFandMORF; see Ghiselli et al., 2013; Milani et al., 2013a) and F-type cytochrome c oxidase subunit 2 duplication (COX2B)have any OXPHOS function, they were not included here.The transcription of both mt and nuOXPHOS genes showed significant differences across tissues (in both cases, Kruskal-Wallis test:P<0.001), with nuOXPHOS genes presenting higher transcription in most pairwise comparisons between adductor muscles and other tissues and mtOXPHOS genes presenting higher transcription in most pairwise comparisons between gonads and somatic tissues (Significance for pairwise comparisons in Supplementary Table S4).Moreover,mtOXPHOS genes showed an overall significantly higher transcription than nuOXPHOS in all the tissues except female mantles (Figure 2).M-type OXPHOS genes presented remarkably higher transcription in male gonads than F-type and nuOXPHOS genes, which is consistent with the deviation of M-type OXPHOS genes in Figure 1.The transcription level for each OXPHOS gene is shown in Supplementary Figure S3(A and B, respectively).Intriguingly, the nuOXPHOS succinate dehydrogenase cytochrome b560 (SDHC) had two divergent sequences (SDHC-1andSDHC-2) and one of them (SDHC-2)presented a gonad-specific transcription (in both males and females).
Strong co-transcription signal within mitochondrial and within nuclear OXPHOS genes across tissues, but weak or absent across genomes
To test the hypothesis that genes involved in the same biochemical activity tend to be co-transcribed inR.philippinarum(Shyamsundar et al., 2005; Stuart et al., 2003),we calculated Spearman‘s rho between pairwise OXPHOS genes.According to the transcriptional correlations across tissues, all OXPHOS genes were clustered into four majordistinct groups: F-type, M-type, nuOXPHOS1 and nuOXPHOS2.Genes from two nuOXPHOS subclusters showed a less pronounced, but still positive correlation between each other (Figure 3A).To further investigate the correlation strength within and between different gene components, we plotted the correlation coefficients (rho) of OXPHOS genes separately for each component, and we randomly selected a subset of nuclear genes as a control to evaluate our observation.We found that the correlation within mtOXPHOS genes (F-type or M-type) is higher than the correlation within the nuOXPHOS, which in turn is higher than the correlation within randomly selected nuclear genes(Figure 3B).The correlation between F-type and nuOXPHOS genes is slightly higher than the correlation between F-type and random nuclear genes, indicating a weak co-transcription signal between F-type and nuOXPHOS genes, while thecorrelation between M-type and nuOXPHOS genes is remarkably higher than the correlation between M-type and random nuclear genes (Figure 3C).Considering that the M-type transcription was primarily detected in male gonads, it is unclear whether the high co-transcription signal between M-type and nuOXPHOS results from a male-gonad specific transcription or reflects actual co-transcription.Therefore, we investigated the correlation between mt and nuOXPHOS genes separately for each tissue.We found that the correlation between M-type and nuOXPHOS genes in male gonads was not significantly different from the correlation between M-type and randomly selected genes (Wilcoxon ranksum test with Bonferroni correction, P>0.05), indicating that the significant co-transcription signal across tissues between M-type and nuOXPHOS genes in Figure 3C was due to the gonad-specific transcription of M-type in male gonads(Supplementary Figure S4).Moreover, the co-transcription signal between F-type and nuOXPHOS genes was not consistent in different tissues (Supplementary Figure S4).Weak co-transcription was detected in the female mantle between F-type and nuOXPHOS genes, but the signal disappeared in other tissues (Supplementary Figure S4).Taken together, our results indicated a weak or absent cotranscription signal between mt and nuOXPHOS genes, but a strong co-transcription signal within F-type, within M-type, and within nuOXPHOS genes.
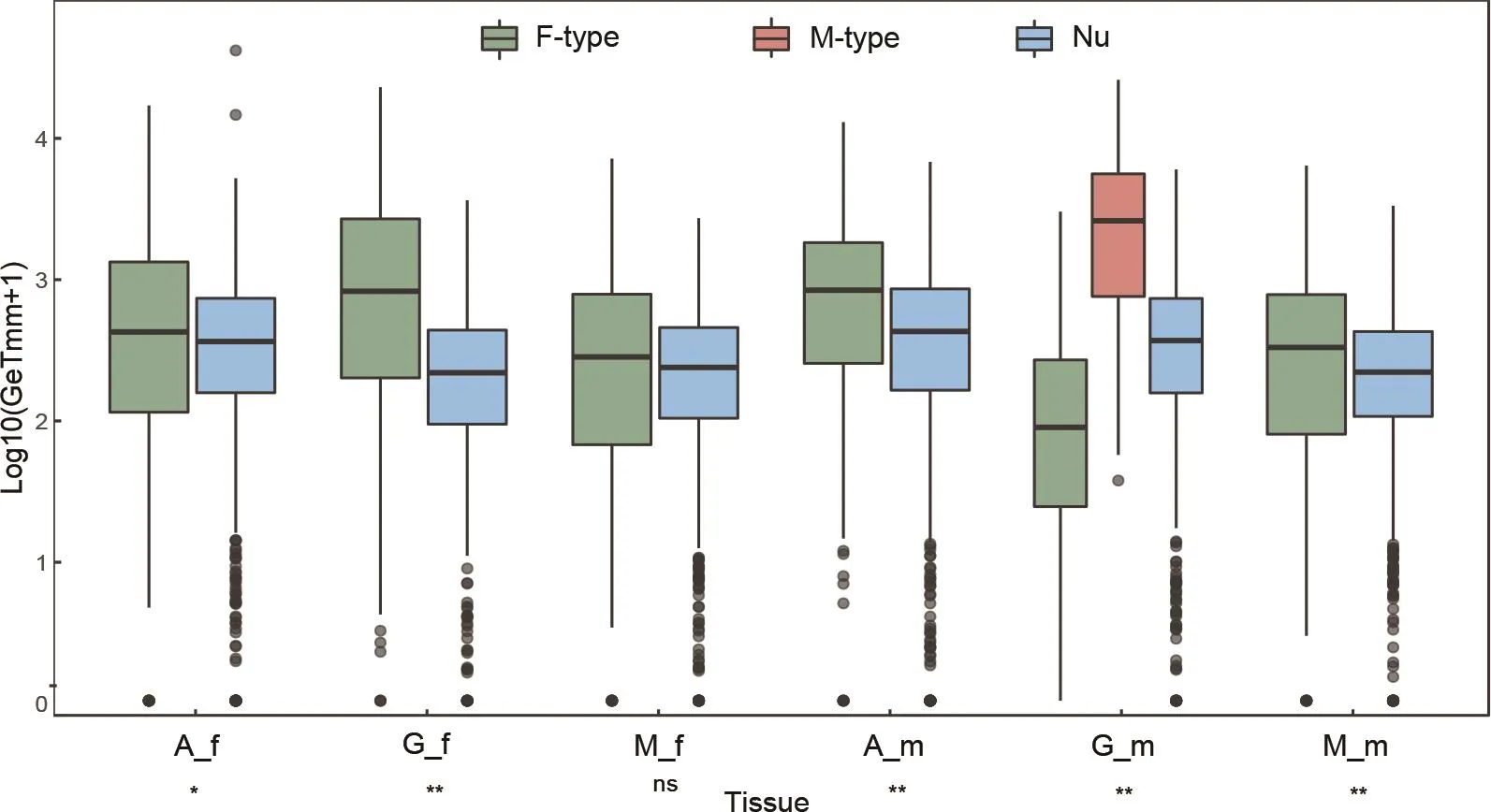
Figure 2 The transcription level of mitochondrial and nuclear OXPHOS genes across tissues

Figure 3 The co-transcription between mitochondrial and nuclear OXPHOS genes
To test the hypothesis that genes within the same complex(intracomplex) presented a stronger correlation than the genes between different complexes (intercomplex) (Garbian et al.,2010; van Waveren & Moraes, 2008), we plotted the correlation coefficient of nuOXPHOS genes separately for each complex and compared them with intercomplex correlation coefficients (“Cinter” in Figure 3D).Such analysis was not performed for mtOXPHOS because of only a few genes being present in each complex.The nuOXPHOS genes belonging to the same complex were not clustered together(Figure 3A) and the correlation coefficients within the same complex were not significantly different from intercomplex correlation coefficients except for complex I and V (Wilcoxon rank-sum test with Bonferroni correction, P<1e-5), in which intracomplex coefficients were slightly higher than the intercomplex coefficients (Figure 3D).
Nuclear genes co-transcribed with OXPHOS genes are enriched for mitochondrial processes
The exceptional heteroplasmic condition in DUI bivalves raises some questions: how is the compatibility between nuOXPHOS and two highly divergent mtDNA populations maintained? And which genes or pathways could be possibly involved in coordinating the OXPHOS process? To address these questions, we retrieved the nuclear genes that were cotranscribed with OXPHOS genes.We plotted the distribution of P-values from Spearman‘s correlation and the corresponding rho for all the co-transcribed nuclear genes(Supplementary Figure S5), and a strict cutoff (rho>0.6) was used to ensure reliability.In this way, a total number of 136,1 077 and 3 468 nuclear genes showed a significantly positive correlation with the F-type, M-type, and nuclear OXPHOS genes, respectively (Supplementary Table S5).Many nuclear genes co-transcribed with mtOXPHOS genes were involved in the assembly of OXPHOS complexes, mitochondrial stability,and quality control.Also, a large number of nuOXPHOS genes were co-transcribed with genes encoding ribosomal proteins and genes involved in the TCA cycle (Supplementary Table S5).To further investigate the function of these co-transcribed nuclear genes, we performed a GO enrichment analysis and the results are shown in Supplementary Table S6.The overrepresented nuclear genes co-transcribed with F-type OXPHOS genes were associated with homeostatic process,mitochondrial respiratory chain complex assembly, and regulation of cellular pH (Supplementary Table S6).On the other hand, the nuclear genes co-transcribed with M-type OXPHOS genes presented a different situation, with reproductive process, nucleotide phosphorylation, and cell cycle being enriched (Supplementary Table S6).The nuclear genes correlated with nuOXPHOS were involved in the biosynthetic process, protein metabolic process,mitochondrion organization, gene expression, and translational initiation (Supplementary Table S6).To identify candidates possibly involved in the transcriptional regulation of mt and nuOXPHOS, we focused on the co-transcribed nuclear genes annotated as transcription factors, or that contain DNA or RNA binding sites.The candidate genes are listed in Supplementary Table S7.
Polymorphism and divergence in OXPHOS genes
The average number of SNPs across all samples identified in F-type, M-type, and nuOXPHOS were 50, 118, and 201,respectively.Figure 4A shows the percentage of synonymous and nonsynonymous SNPs in the different gene components,with F-type presenting a significantly higher percentage of nonsynonymous SNPs, M-type and nuOXPHOS genes presenting a higher percentage of synonymous SNPs(Figure 4A; Supplementary Table S8).However, a high percentage of nonsynonymous SNPs were found in one COX2 copy, named COX2B, in the F-type (Supplementary Figure S6A).If we exclude COX2B, the percentage of synonymous and nonsynonymous SNPs in F-type OXPHOS genes were not significantly different from the respective categories in the M-type (Figure 4A; Supplementary Table S8).Ghiselli et al.(2013) observed a markedly different transcription level between the two COX2 copies, with COX2B showing a lower transcription.They hypothesized that COX2B might be undergoing a pseudogenization process.The high number of nonsynonymous variants in COX2B resulting from this work is consistent with such a hypothesis.The relative ratio of synonymous and nonsynonymous SNPs in mt and nuOXPHOS genes are shown in Supplementary Figure S6A, B.
To assess patterns of selection in OXPHOS genes, we applied two approaches: frequency spectrum-based test, and McDonald-Kreitman (MK) test.Allele frequency was calculated for three different components of OXPHOS genes and a set of randomly selected genes.Four distinct distributions were observed: one for the F-type in gonads (note that the distributions for the F-type in female and male gonads were not significantly different from each other), one for the M-type,one for random genes in gonads, and one for nuOXPHOSgenes in gonads (Kolmogorov-Smirnov test with Bonferroni correction:P<0.001, Figure 4B).M-type OXPHOS genes presented a remarkably high intermediate allele frequency.Tajima‘sD, Fu & Li‘sDand Fu & Li‘sFshowed negative values for most F-type, nuOXPHOS genes, and randomly selected nuclear genes, but positive values for most M-type OXPHOS genes (Supplementary Table S9).

Figure 4 The SNP effects and allele frequency in OXPHOS genes
For mtOXPHOS genes, we firstly compared the polymorphism (Pn and Ps) and divergence (Dn and Ds)between F-type and M-type OXPHOS genes.As shown in Supplementary Table S9, except for theATP8,COX3, NAD3 and NAD4L,all the rest of mtOXPHOS genes showed significant neutrality index (NI).NI is derived from the MK test and it quantifies the direction and degree of departure from the neutrality: NI=1 indicates neutrality; NI>1 indicates negative selection; NI<1 indicates positive selection (Rand & Kann,1996).Therefore, most mtOXPHOS genes showed a signal of positive selection between F-type and M-type.We also used the direction of selection (DOS) to evaluate the data in the MK test.The positive value of DOS could be consistent with positive selection, whereas the negative value indicates the presence of slightly deleterious mutations segregating in the population (James et al., 2016; Stoletzki & Eyre-Walker,2011).Similarly, the DOS test also indicated possible positive selection in most mtOXPHOS genes.To test if the positive selection signal is present in the F-type or M-type OXPHOS genes or both, we also performed the MK test usingR.decussatus(SMI species) as an outgroup.Interestingly, most F-type OXPHOS genes showed extremely low polymorphic differences which yield the excess of non-significant NI, while most M-type OXPHOS genes presented relatively high polymorphic differences and significant NI<1, which could be consistent with positive selection acting on M-type OXPHOS genes (Figure 5A; Supplementary Table S9).The MK test was also performed on nuOXPHOS and randomly selected nuclear genes.A considerable number of nuOXPHOS and randomly selected nuclear genes presented non-significant NI and a nearly equal ratio of polymorphic and divergent differences,consistent with neutrality (Figure 5B; Supplementary Table S9).However, a large proportion of nuOXPHOS genes and some randomly selected genes also presented relatively high divergence, indicating the signature of positive selection.
To test whether nuOXPHOS subunits that are predicted to be in contact with mtOXPHOS subunits presented a different percentage of synonymous and nonsynonymous SNPs, we divided the nuOXPHOS genes into two groups (see Piccinini et al., 2021 for details): the “contact” group which was supposed to physically contact mtOXPHOS subunits; and the“non-contact” group which was predicted to have no direct interaction with mtOXPHOS subunits.Although the contact group presented a higher percentage of both synonymous and nonsynonymous SNPs than the non-contact group(Supplementary Figure S6C), the MK test indicated that NI index in contact and non-contact groups were not significantly different from each other (Wilcoxon rank-sum test:P>0.05;Supplementary Table S9).
Codon usage bias in OXPHOS genes
Mt and nuOXPHOS genes showed remarkably different GC compositions (Table 1), with extremely high AT skew in mtOXPHOS, as also found in other eukaryotes.Interestingly,we also found significant differences in GC composition between F- and M-type OXPHOS genes.F-type and M-type OXPHOS genes showed a similar percentage of GC12 (F-type: 34.42%; M-type: 34.83%), while a significantly higher percentage of GC3 was found in M-type (F-type: 21.69%; M-type: 26.91%) (Table 1; Supplementary Figure S7).Moreover,M-type OXPHOS genes presented generally slightly higher ENC and lower CAI values compared to F-type (ENC:P>0.05;CAI:P<0.05; Table 1; Supplementary Figure S7), indicating alleviated CUB in M-type OXPHOS genes.NuOXPHOS genes displayed relatively high ENC and CAI values that are comparable to those of the other nuclear genes in the transcriptome (Wilcoxon rank-sum test with Bonferroni correction,P>0.05).Although under different CUB, the heatmap based on RSCU values revealed that both mt and nuOXPHOS presented a shared usage bias towards A/U-ending codons (Supplementary Figure S8).
ENC and GC3 relation plot (Nc-plot) compares the actual distribution of genes to an expected distribution which assumes no selection, therefore the departure from theexpected curve indicates that these genes are under selective pressure (Wright, 1990).Likewise, the neutrality test plots the GC12 against GC3 to reflect the equilibrium between mutation pressure and natural selection (Sueoka, 1999).In this study,the Nc-plot revealed that only a small number of OXPHOS genes laid on the expected Nc curve, with most genes departing from the corresponding predictions (Supplementary Figure S9).The neutrality test showed no correlation between GC12 and GC3 in both mt and nuOXPHOS (Table 1),indicating that mutation bias was not the only factor affecting CUB.Negative correlation between CAI and ENC was observed in M-type OXPHOS genes.CAI indicates the selection for the preferred codon, while ENC is a nondirectional parameter for either selection or mutation bias,therefore the correlation between CAI and ENC could indicate the role of selection, but mutation would lessen this correlation(Behura & Severson, 2012).Significant negative correlations between GC3 and CAI were observed in both F-type and M-type OXPHOS genes (Table 1), indicating that GC3 may be associated with CUB in mtOXPHOS genes.Moreover, GCeq in both F-type and M-type OXPHOS was slightly higher than GC3, while GCeq in nuOXPHOS is lower than the GC3(Table 1).All these tests indicated that mutation alone cannot explain the CUB in OXPHOS genes.In nuOXPHOS genes, a significant correlation between CAI and transcription level was observed, indicating the possible role of translational selection.Similarly, for the protein-coding genes in the transcriptome, significant correlations were also detected between GC3 and CUB, and between CAI and transcription.

Table 1 Statistics for codon usage in different OXPHOS gene components

Figure 5 The results for McDonald-Kreitman test
The COA analysis based on the codon counts indicated that Axis 1 was significantly correlated with the CAI and GC3 in both M-type and nuOXPHOS genes, while no such correlation was observed in F-type OXPHOS genes (Figure 6; Table 1).Besides, the correlation between transcription and Axis 1, and between transcription and Axis 2 were observed in three different OXPHOS components.The COA analysis based on RSCU also showed similar results (Table 1).Consistently,these results indicated the GC3 composition and transcription levels could be responsible for the CUB for both OXPHOS genes and protein-coding genes in the transcriptome.Moreover, the chi-square test for the context-dependent mutations in F-type, M-type, and nuOXPHOs genes all indicated that the third position bases were not independent of the second position bases (Supplementary Table S10, F: X-square=143.25,P<0.001; M: X-square=111.2,P<0.001;NuOXPHOS: X-square=328.08,P<0.001).
DlSCUSSlON
Distinct transcriptional dynamics and regulatory mechanism in OXPHOS genes
Although the quantification of mtDNA and mtRNA in DUI species has been investigated before (Breton et al., 2017;Dalziel & Stewart, 2002; Ghiselli et al., 2011; Milani et al.,2014; Obata et al., 2011), the transcription patterns in DUI species across tissues in both sexes are still largely unknown.In the present study, we assessed the transcription of F-type and M-type genes in adductors, mantles, and gonads of a DUI speciesR.philippinarum.We found that the transcription levels of F-type OXPHOS genes were significantly different across tissues, while M-type OXPHOS genes were highly transcribed only in male gonads (Figure 2).Traces of M-type mtRNA (>1% reads mapped to M-type in the sample) were also detected in 5 (out of 28) male somatic samples analyzed(Supplementary Figure S1 and Table S2).The presence of M-type mtRNA in somatic tissues seems to vary across DUI species, for example 60% male somatic samples inU.peninsularisand 89.5% male somatic samples inV.ellipsiformispresented somatic transcription of M-type mt genes (Breton et al., 2017).InR.philippinarum, previous work reported variable number of M-type DNA and RNA in somatic tissues, depending on the individual (Ghiselli et al., 2011;Iannello et al., 2021; Milani et al., 2014).In this work, we found that M-type is barely transcribed in the somatic tissues of most males.Such a pattern could be due to a low number of M-type mtDNA in these samples or tissue-specific transcription of M-type in males, which leads the M-type to be highly transcribed in male gonads, but poorly transcribed in male somatic tissues.Ghiselli et al.(2011) detected M-type mtDNA in most of (~87%) male somatic tissues, and Milani et al.(2014)reported no correlation between the transcription level of cytochromeb(cytb) and its DNA copy number in males ofR.philippinarum.These evidences were in line with a tissuespecific regulation of transcription of M-type, which is independent of the M-type DNA copy number.On the other hand, the extremely low transcription levels observed in somatic tissues of many males could reflect an absence of M-type mtDNA in such samples.Future investigations including sequencing of both mtDNA and mtRNA will help to clarify this point.

Figure 6 The relationship between codon usage and GC3 (A), CAl (B) and transcription (C, D)
Transcription levels clearly distinguished F-type, M-type,and nuOXPHOS genes into three groups (Figure 1), indicating distinct transcription dynamics.Tissue-specific transcription was observed in F-type and nuOXPHOS genes, with nuOXPHOS genes presenting higher transcription in adductors and mtOXPHOS genes presenting higher transcription in gonads (Figure 2; Supplementary Table S4).Similar tissue-specific transcription of OXPHOS was widely reported and quantified in humans and mice, and this transcription pattern was proposed to be associated with differences in metabolic profiles and variable energetic demands in different tissues (Barshad et al., 2018; van Waveren & Moraes, 2008).However, different from the results in van Waveren & Moraes (2008), which showed stronger correlations within than among complexes—in this study we observed slightly higher intracomplexes correlation only in complex I and V (Figure 3D).This discrepancy may be explained by several reasons: (1) In DUI species, nuclear OXPHOS genes must cooperate with two mitochondrial genomes, which might loosen the correlations between different complexes; (2) R.philippinarum is a sedentary living bivalve that has lower energy needs, therefore it may be subject to weaker selection in maintaining OXPHOS processes compared to the other taxa (Piccinini et al., 2021;Iannello et al., 2019); (3) Normalization methods may also influence co-transcription results, and recent study on large human dataset indicated that normalization techniques based on total read count (such as TPM or FPKM) may lead to artefactual positive correlations (Perez & Sarkies, 2021).
Significant positive co-transcription was observed separately in F-type, in M-type, and in nuOXPHOS genes, but co-transcription signal between mtOXPHOS and nuOXPHOS was weak/absent and not consistent across tissues (Figure 3;Supplementary Figure S4).Considering the distinct transcriptional trends and the separate co-transcription patterns, our data indicate that the transcriptional difference of nuOXPHOS genes was independent of the M- or F-type genome and that the transcription of F-type, M-type, and nuOXPHOS genes might be under different regulatory mechanisms, including co-translational regulation (Couvillion et al., 2016).
Nuclear OXPHOS genes were subdivided into two positively correlated clusters (Figure 3).While the reason behind this split is unknown, one possibility could be the presence of supercomplexes within nuOXPHOS genes, with a tighter coregulation inside each.Several different types of supercomplexes (such as CI+CIII+CIV, CIII+CV, CI+CIII) have been established in model organisms and several genes(COX7, COX6A, NDUFB4, NDUFB9, UQCRC1, UQCRQ) are involved in supercomplex formation (reviewed in Chaban et al., 2014; Milenkovic et al., 2017).However, the mechanisms and functional roles of supercomplexes are still largely unknown, especially in non-model organisms.
Candidate pathways and genes associated with OXPHOS co-transcription
In the present study, the mt and nuOXPHOS genes were cotranscribed with nu-encoded genes involved in many biological processes, such as mitochondrial respiratory chain complex assembly, cellular homeostasis, and translation(Supplementary Table S6).Mitochondrial respiratory chain complex assembly is an intricate process that requires tightly orchestrated co-regulation from both mitochondrial and nuclear genomes (Tang et al., 2020).Different nu-encoded assembly factors were observed to be co-transcribed with mt and nuOXPHOS genes (Supplementary Table S5) and these factors have been previously shown to be essential for the proper assembly and function of the OXPHOS system (van Waveren & Moraes, 2008).Genes involved in cellular homeostasis were overrepresented for genes co-transcribed with the F-type OXPHOS genes, along with many genes involved in mitophagy and ubiquitination processes(Supplementary Tables S5, S6).This is in line with our expectations, as mitochondria are also essential for cellular homeostasis, calcium signaling, and metabolite synthesis.The proper function of mitochondria also requires balanced coordination between mitochondrial biogenesis and mitophagy through complex signaling pathways (Willems et al., 2015).
Notably, the translation process was overrepresented for genes co-transcribed with nuOXPHOS genes, indicating that nuOXPHOS genes might also be under translational coregulation (Supplementary Table S6).By contrast, nuencoded genes co-transcribed with M-type OXPHOS genes were overrepresented in the reproductive process and spermatogenesis (Supplementary Table S6).One reason could be that the M-type OXPHOS genes were found almost only in male gonads among our samples and thus the nuclear genes co-transcribed with M-type OXPHOS genes might also co-express with the genes responsible for the development of male gonads.Alternatively, M-type OXPHOS genes might be directly associated with reproduction in DUI species.According to previous studies, M-type mitochondria might be involved in some aspects of sex differentiation in DUI species as suggested by several authors (Ghiselli et al., 2012; Milani et al., 2013b; Zouros, 2013).Since M-type is limited to male gonads in these samples, it is not surprising to see the cotranscription between gonad-specific genes and M-type OXPHOS genes.Therefore, co-transcription could reflect either functional interaction between M-type and gonadspecific nuclear genes, or similar independent transcription profiles.
It is worth mentioning that Maeda et al.(2021) found two divergent SDHC sequences in their study and one of them(SDHC-2) showed a gonad-specific transcription(Supplementary Figure S3B).Around half of the nuclear genes co-transcribed with nuOXPHOS were correlated only with the SDHC-2, which might explain the presence of enriched GO terms involved in the cell cycle (Supplementary Table S6).Indeed, SDHC is important for both OXPHOS and Krebs cycle, and studies indicated that deficiency in this gene wouldincrease ROS production and induce metabolic stress,genomic instability, and hypoxia (Slane et al., 2006; Tretter et al., 2016).However, the reason behind this remarkable tissuespecific expression is not clear.A recent study in humans indicated that tissue-dependent splice variants and OXPHOS subunit paralogs may also be involved in retaining OXPHOS activities (Barshad et al., 2018).
Considering the strong co-transcription within F-type, within M-type and within nuOXPHOS genes, it is reasonable to speculate that nuclear regulators may be responsible for transcribing each group.Here we identified a group of candidate regulator genes that are either transcription factors or had a DNA/RNA binding domain (Supplementary Table S7).These genes included zinc finger protein 341, which can activate transcription factor STAT1, a gene previously shown to regulate the OXPHOS process (Pitroda et al., 2009).Pentatricopeptide repeat domain-containing protein 3 is a mitochondrial RNA-binding protein and proteins containing PPR domains are known to play a role in transcription, RNA processing, splicing, stability, editing, and translation (Miglani et al., 2021; Schmitz-Linneweber & Small, 2008).Moreover,the roles of PPR proteins in mitochondrial gene expression and OXPHOS process has also been reported (Lightowlers &Chrzanowska-Lightowlers, 2008; Soto et al., 2021).Recently,a ribosome profiling study in human cells revealed that balanced mito-nuclear OXPHOS synthesis requires a nuclearencoded mt protein LRPPRC (Soto et al., 2021).
Selection acts on both nonsynonymous and synonymous sites of OXPHOS genes
In DUI species, interspecific comparisons have found that M-type genes accumulate more mutations and have a higher evolutionary rate than F-type (Hoeh et al., 1996, 1997, 2002;Liu et al., 1996; Ort & Pogson, 2007; Śmietanka et al., 2009;Soroka & Burzyński, 2010; Stewart et al., 1995, 1996;Zbawicka et al., 2010), which led to the hypothesis that F- and M-type genomes experienced different selective pressures(reviewed in Zouros, 2013).Here, we found that MK tests between F-type and M-type in R.philippinarum, and between mt genes in R.decussatus and M -type in R.philippinarum genes consistently indicated positive selection on most M-type OXPHOS genes (Figure 5).M-type COX3 and NAD3 did not show departure from the neutral expectations, indicating that selection on M-type might be variable in different genes.On the other hand, intermediate allele frequency and positive Tajima‘s D in M-type OXPHOS genes suggested the possibility of population bottleneck or balancing selection(Figure 4).Native to the Pacific coast of east Asia, R.philippinarum was first transported to America in the 1930s,and then was transferred to Europe to cope with the production decline of local clam species during 1970s–1980s(Chiesa et al., 2017; Cordero et al., 2017).Several researches revealed that reduced genetic diversity and genetic differentiation compared to the American population were consistent with a strong founder effect in the European population (Chiesa et al., 2017; Cordero et al., 2017).Thus, in line with these studies, the founder effect can also explain why F-type and nuclear genes presented generally negative Tajima‘s D and excess of rare alleles.
However, the founder effect was not sufficient to explain why M-type showed such different patterns both in the Italian population and also in the American population (Ghiselli et al.,2013).One possibility could be the narrower germline bottleneck of M-type mtDNA.Past studies indicated that the F-type mtDNA copy number in eggs is on average 10 times higher than the copy number of M-type mtDNA in sperm(Ghiselli et al., 2011), and that the narrower genetic bottleneck in M-type mtDNA could lead to the segregation of mtDNA variants in different tissues, causing remarkable withinindividual variation and therefore also higher variability between samples (Iannello et al., 2021).Alternatively, the presence of high intermediate frequency alleles and positive Tajima‘s D could be a signal of balancing selection.Balancing selection on mtDNA has been found in gynodioecious plants showing cytoplasmic male sterility (CMS), in which a population consisting of both females and hermaphrodites and the sex is determined by the interaction between mitochondrial male-sterility genes and nuclear restorer-of-fertility genes(reviewed in Chase, 2007; Delph & Kelly, 2014).Under balancing selection, restorer genes are not fixed in the population because of the “cost of restoration” and CMS genes are under negative frequency-dependent selection to maintain the long-term balanced sex ratio in the population(Delph & Kelly 2014).DUI and CMS show some common features (Breton et al., 2010, 2011; Ghiselli et al., 2013; Milani et al., 2016; Mitchell et al., 2016): (1) the presence of novel lineage-specific mt-ORFs (Ghiselli et al., 2013; Milani et al.,2013a; Mitchell et al., 2016), which allows potential interaction between mitochondria and nuclear genes; (2) an excess of mid-frequency polymorphism in M-type mtDNA (Ort & Pogson,2007; Quesada et al., 1998), which might lead to match/mismatch between mitotype and nuclear genes; (3) the hypothesized association between mtDNA and sex determination in DUI species (reviewed in Breton et al., 2017;Zouros, 2013); (4) the presence of biased sex ratios (the proportion of males in populations range from 8% to 83%)(Ghiselli et al., 2012; Yusa et al., 2013); (5) recombination of M-type mtDNA in male gonads (Burzyński et al., 2003;Ladoukakis & Zouros, 2001), which allows for the emergence of divergent mitotypes.Under balancing selection, M-type polymorphisms in the population will not be fixed because two major mitotypes might have different fitness to the environment and mitotypes might interact with the nuclear genes to determine/differentiate the sex.Certainly, this is just speculation and more studies are needed to shed light on such aspects.
It is worth mentioning that positive selection on M-type has also been reported in other DUI species (Ort & Pogson, 2007;Śmietanka et al., 2009), and that selection on M-type inferred by population genetic tests (e.g., McDonald-Kreitman test) and phylogeny-based method (e.g., dN/dS) have been inconsistent in many cases (Ort & Pogson, 2007; Śmietanka et al., 2009;Zbawicka et al., 2010).In R.philippinarum, phylogenetic analysis indicated relaxed selection on most M-type OXPHOS genes (Maeda et al., 2021), whereas population tests in this study showed a signal of positive selection or balancing selection.Although demographic events or bottleneck differences may influence population-based methods, severalgenes such as COX3 and NAD3 showed the same signal between the two methods.Moreover, these two genes showed different signals compared to the other genes in population-based tests (see discussion above, Supplementary Table S9), which cannot be explained by demographic events because we should see the same signal across all genes if demographic changes were the major forces.
Selection on synonymous codon usage in different components of OXPHOS genes was also detected.Although mt genomes usually encode only one tRNA for each codon family, more and more evidence from numerous organisms indicated that pools of tRNA in mitochondria include both locally encoded and imported tRNAs (see Rubio & Hopper,2011; Salinas-Giegé et al., 2015 for a review), which enables selection for both efficiency and accuracy on mt genes.The deviation from the expected ENC values in the Nc plot(Supplementary Figure S9) and lack of correlation between GC12 and GC3 (Table 1) indicated a role of natural selection in CUB.Moreover, the significant and high correlation between transcription level and CUB may reflect the selective pressure to optimize the codon usage in highly transcribed mRNA to avoid sequestration of ribosomes and slow down the elongation rate (Gingold & Pilpel, 2011; Plotkin & Kudla,2011).Similarly, translational selection was also detected in nuclear OXPHOS genes (Table 1; Figure 6).However,translational selection in mt and nuOXPHOS seems to favor codons with different endings.In both mt and nuOXPHOS genes, GC3 showed a significant impact on codon usage.Whereas GC3 in mtOXPHOS genes was lower than GCeq,GC3 in nuOXPHOS genes was higher than Gceq, suggesting that translational selection in AT-rich mt genomes drives the codons into an A/U ending, while selection in nuOXPHOS genes drives the codons into G/C ending.Despite the presence of translational selection in OXPHOS genes, we argue that the mutational bias is still a major force in OXPHOS genes.Our results revealed that CUB in OXPHOS genes is shaped by the balance between selection favoring preferred codons and mutation bias coupled with random drift.
The coordination of OXPHOS genes may involve translational regulation
Although selection on silent sites does not result in changes to the protein sequence, it can still drive protein evolution in terms of expression regulation.With the advent of highthroughput sequencing, increasing evidence shows that translational selection on CUB facilitates the regulation of gene expression and the generation of differential protein abundance (Camiolo et al., 2012; Horn, 2008; Jeacock et al.,2018; Najafabadi et al., 2009).It was hypothesized that codon usage may be selected during evolution to synchronize the efficiency of translation with functional requirements for the expression of specific proteins at certain times, in a specific tissue (Camiolo et al., 2012; Najafabadi et al., 2009).
The tight interaction between mt and nuOXPHOS requires the coordinated regulation of gene expression to ensure the demands for cellular energy are met.In the present study, we found separate co-transcription within F-type, M-type, and nuOXPHOS genes but weak or absent co-transcription between OXPHOS genes across genomes (Figure 3;Supplementary Figure S4), suggesting that co-regulation of OXPHOS genes is not at transcription stage.Instead,translational selection was detected for both mt and nuOXHPOS genes (Table 1; Figure 6), suggesting the possibility of co-regulation at the translation level.Although translational selection was detected in all three different components of OXPHOS genes, the selective strength seems to be different.A significant correlation was detected between CAI and transcription in nuOXPHOS genes and also weakly in nuclear protein-coding genes, but not in mtOXPHOS genes,indicating translational selection in nuOXPHOS genes may be stronger than mtOXPHOS genes.Selection on F- and M-type mt genes may also be different.Translational selection might be stronger in M-type, indicated by the significant positive correlation between CAI and axis 1 in correspondence analysis and significant negative correlation between CAI and ENC.Therefore, our results are consistent with previous hypotheses that translation in mt genes is less efficient than nuclear genes (Adrion et al., 2016; Havird & Sloan, 2016; Pett& Lavrov, 2015; Sloan et al., 2013; Woodson & Chory, 2008).Combined with previous studies in yeast and humans that indicated translational regulation during OXPHOS complex synthesis (Couvillion et al., 2016; Soto et al., 2021), we speculate that the different strengths of translational selection on OXPHOS genes may be responsible for regulating protein abundance of OXPHOS genes and that the coordination of expression of OXPHOS genes may involve translational regulation in DUI species.
CONCLUSlONS
In addition to the common knowledge of co-transcriptional coordination between mt and nuOXPHOS genes in mammals,our study revealed that coordination in other species,particularly in DUI species, could be different and might involve post-transcriptional/translational regulation.We found a clear co-transcription signal within F-type, within M-type and within nuOXPHOS genes, but the signal is weak or absent between mt and nuOXPHOS genes, suggesting that coordination between mt and nuOXPHOS genes may not occur at the transcription level in DUI species.It will be interesting to assess if such situation is due to a peculiarity of the DUI system, or if it is more widespread across bivalves and/or other invertebrates.Translational selection on synonymous codon usage of both mt and nuOXPHOS genes further indicated the possible role of translational regulation in coordinating the OXPHOS genes.Together, these results advance our understanding of the coordination between mt and nuOXPHOS gene, and provide a new perspective of diverse and complex coordination mechanisms of OXPHOS genes in the animal world.
DATA AVAlLABlLlTY
The raw reads of Ruditapes philippinarum were deposited in the National Center for Biotechnology Information (NCBI)database under BioProjectID PRJNA672267 (SRA accession No.SRR12904870–SRR12904958).The assembled transcripts were deposited in the Transcriptome Shotgun Assembly Sequence (TSA) database of NCBI (accession No.GIVW00000000).
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETlNG lNTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRlBUTlONS
F.G.and L.M.obtained the samples for this study.R.X., M.I.,F.G., and J.C.H.designed the study.R.X.performed the analysis and wrote the manuscript.F.G., M.I., L.M., and J.C.H revised the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Giovanni Piccinini for providing the sequences of OXPHOS genes in R.decussatus, and two anonymous reviewers for their insightful comments on early versions of the manuscript.
杂志排行

Zoological Research的其它文章
- Zoological Research shines in the East
- Cenozoic Tethyan changes dominated Eurasian animal evolution and diversity patterns
- Genome-wide association study identifies quantitative trait loci affecting cattle temperament
- Opah (Lampris megalopsis) genome sheds light on the evolution of aquatic endothermy
- Comparative mitogenomic analyses unveil conserved and variable mitogenomic features and phylogeny of Chedrinae fish
- A new species of the gudgeon genus Huigobio Fang,1938 (Cypriniformes: Cyprinidae) from the Yangtze River Basin, southern China