1例德朗热综合征患儿的NIPBL基因突变分析
2022-01-26徐瑞张英懿刘心洁
徐瑞,张英懿,刘心洁
1山东大学齐鲁医院儿科,济南250012;2寿光市人民医院儿科
德朗热综合征(CdLS)是一种以严重神经发育障碍为主要表现的遗传综合征,其在活产患儿中的发病率为1/10 000~1/30 000,主要为散发性,极少为家族聚集性发生。CdLS的临床表现包括智力发育落后、特殊面容、宫内和生后生长迟缓及多脏器畸形等[1-3]。此外,有报道显示,CdLS患儿体液免疫受到影响,较普通儿童更易罹患感染性疾病[4]。目前临床上CdLS经典的致病基因有5种:NIPBL、SMC1A、SMC3、RAD21和HDAC8[5],随着基因检测的普及及检测水平的提高,有更多的致病基因被发现。2020年8月,我们临床确诊1例CdLS患儿,为明确病因,应用高通量捕获测序进行致病基因变异检测,并用Sanger测序验证测序结果以及致病基因的家系分析。现报告如下。
1 资料与方法
1.1 病例介绍 患儿女,2个月又23 d,因“发现头围偏小1 d”就诊于我院门诊。患儿系第1胎第1产,孕38+6周经阴道分娩,出生过程顺利,出生体质量3 400 g,生后无窒息缺氧病史。出生后听性脑干反应未通过,出生第42天复查通过。可追视距离眼前20 cm左右的黑白卡。无相关家族史。体格检查:体质量4 150 g,头围33.7 cm(<-3 SD),前囟仅容指尖。多毛,眉毛浓密、长,后发际线低,耳廓边缘及背部毛发多。长人中,短鼻,口角下歪,上唇薄,小下颌,高腭弓。胸廓无畸形,心肺无异常。躯干有两处2 cm×1 cm咖啡斑,脚趾细长,无通贯掌。四肢肌力、肌张力可,生理反射正常存在,双侧巴氏征阴性。其母孕期第16周行唐氏筛查无异常,孕后期多次彩色超声示胎儿宫内发育迟缓,双顶径<2 SD,家属拒绝行羊水穿刺行基因检查。实验室检查:患儿近期血常规、胸片、肝肾功能+心肌酶+生化、甲状腺功能、血氨未见异常。血串联质谱和尿气相色谱—质谱联用法未见异常。染色体微阵列(CNV)扫描分析未见异常(见OSID码图1)。
1.2 CdLS的判断 根据2020年CdLS国际共识提出的临床特征评分[6],分为基本特征(2分/项)和提示性特征(1分/项)。基本特征包括:①连眉和(或)浓眉;②短鼻、凹鼻嵴和(或)鼻孔前倾;③长人中和(或)人中扁平;④薄上唇红和(或)嘴角下弯;⑤少指(趾)畸形和(或)先天性无指;⑥先天性膈疝。提示性特征包括:①全面发育迟缓和(或)智力障碍;②胎儿宫内发育迟缓(<-2 SD);③出生后生长迟缓(<-2 SD);④小头畸形[产前和(或)产后];⑤小手和(或)短足;⑥第5指发育不全或不发育;⑦多毛。临床特征评分≥11分,且包括至少3个基本特征者诊断为典型CdLS;临床特征评分9~10分,且包括至少2个基本特征者诊断为非典型CdLS;临床特征评分4~8分,且包括至少1个基本特征者,需结合分子测试结果进行诊断;临床特征评分<4分者不诊断CdLS。
1.3 患儿及其父母全基因组检测 经患儿监护人知情同意并签署知情同意书后,抽取患儿及其父母外周血各2 mL,EDTA抗凝,使用QIAamp全血DNA提取试剂盒(德国Qiagen公司)提取基因组DNA。应用GenCap液相捕获目标基因技术(北京迈基诺公司),捕获与CdLS相关的包含CdLS相关基因(NIPBL、SMC3、RAD21、SMC1A和HDAC8)的编码外显子区域和侧翼区域。利用Illumina Nextseq 500第二代测序仪捕获到的区域进行双端测序,读长为150 bp。目标区域测序后,去除测序数据中的接头和低质量数据。运用BWA软件比对到参考基因组上(hg19版本),对测序深度、均一性、探针特异性等数据进行统计分析。
使用GATK软件对该样本的比对数据进行多态性位点检测,对单核苷酸多态性(SNPs)和插入缺失突变(InDels)等数据进行统计和分析,查找SNPs及InDels在千人基因组数据库、人类基因突变数据库、Clinvar数据库、ESP6500外显子测序计划数据库、ExAC数据库(ALL和EAS)及迈基诺内部1 000例正常汉族人群数据库频率,利用SIFT、PolyPhen2、MutationTaster、GERP+等数据库对SNPs及InDels的致病性进行预测分析,筛选上述数据库中频率<0.05且预测结果均为致病性的位点作为与疾病相关的位点。
根据需要测序的DNA片段合成引物,用聚合酶链反应(PCR)法进行扩增,用ABI3730xl测序仪(美国Applied Biosystems公司)以Sanger测序法进行测序,将测序结果与目标区域捕获测序后的结果进行比对。
2 结果
2.1 患儿CdLS临床特征评分结果 患儿CdLS临床特征评分11分,且包括4个基本特征,符合典型CdLS的诊断标准。
2.2 患儿及其父母全基因组检测结果 全外显子基因组测序检出患儿NIPBL基因第6外显子存在c.461G>A杂合错义突变。NIPBL基因c.461G>A变异在各基因数据库及近期文献中均无该位点的相关报道,该位点变异为未报道过的新变异。生物信息学蛋白功能综合性预测软件REVEL预测结果为潜在有害,SIFT、MutationTaster、GERP+预测结果为有害、有害、有害。
经Sanger测序验证,该变异存在于患儿及患儿父亲,c.461G>A可导致编码蛋白第154位精氨酸替代为谷氨酰胺(p.R154Q),见图1。经家系验证分析,其父该位点为杂合突变,其母该位点无变异,其父手臂处多毛,无特殊面容、智力发育落后、生长发育落后、多脏器畸形等表现。
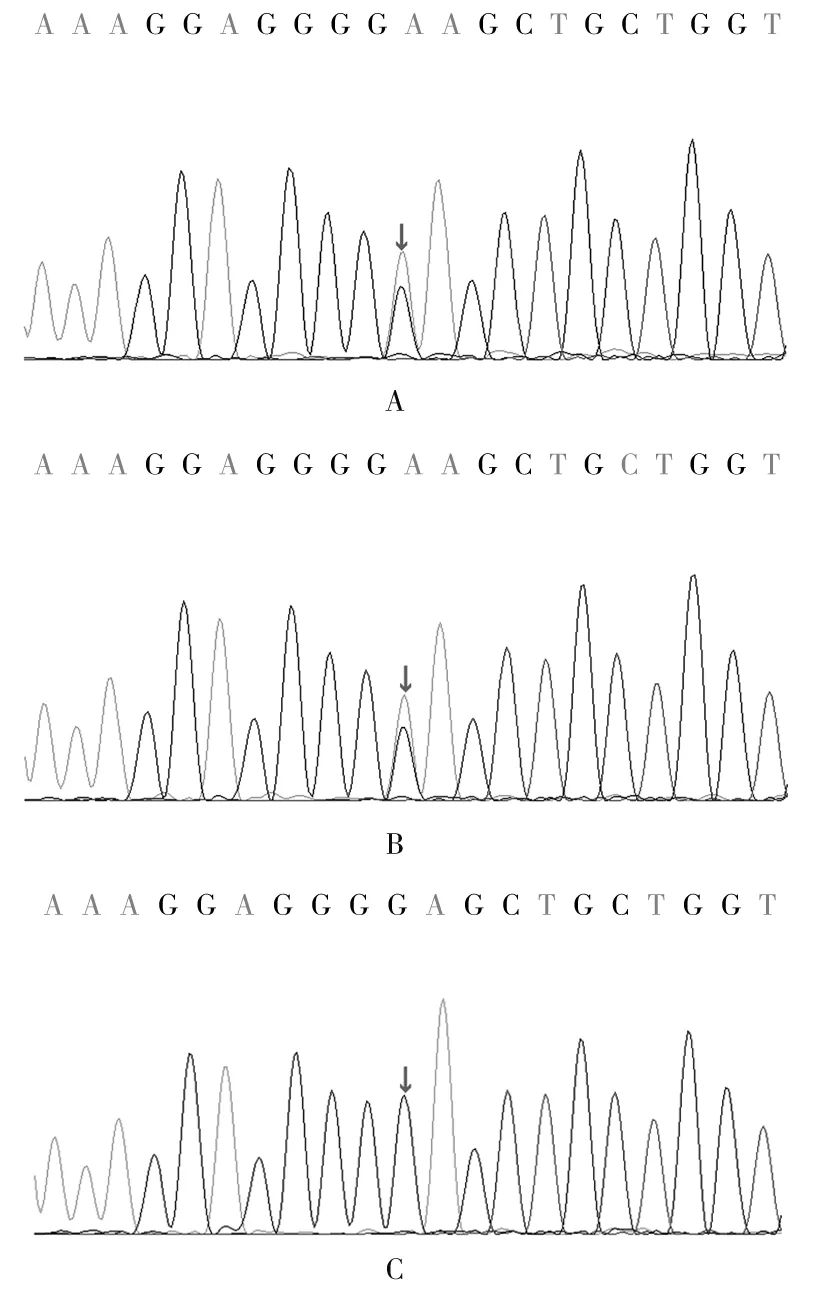
注:A为患儿,箭头所指位置有A与G两种核糖核苷酸,存在杂合变异;B为患儿父亲,箭头所指位置有A与G两种核糖核苷酸,存在杂合变异;C为患儿母亲,箭头所指位置有G一种核糖核苷酸,无变异。
3 讨论
CdLS是一种以严重神经发育障碍为主要表现的遗传综合征,多数为散发病例,<1%的患者有同样受累的父母[6]。目前其诊断主要根据2020年CdLS国际共识提出的临床特征评分,本例患儿宫内发育迟缓(1分),具有典型的特殊面容,如小头畸形(1分)、前发际线低、浓眉(2分)、长睫毛、鼻梁凹陷(2分)、宽鼻尖、长且扁平的人中(2分)、薄上唇红、嘴角下弯(2分)、多毛症(1分),总评分共11分,且包括4个基本特征,可确诊为典型的CdLS。
患儿因生后查体发现头围偏小就诊于我院门诊,经门诊再次详细查体评估后临床诊断为典型CdLS,为进一步验证临床诊断,故行相关基因检测。全外显子基因组测序结果显示,该患儿NIPBL基因第6外显子存在c.461G>A杂合错义突变;其父该位点为杂合突变,其父体征上除手臂出多毛外无特殊面容、生长发育落后表现;其母该位点无变异,为野生型。为验证该突变位点与患儿病症的关系,经生物信息学蛋白功能综合性预测软件REVEL分析,预测结果为潜在有害,SIFT、MutationTaster、GERP+预测结果均为有害。因此推测,NIPBL基因c461G>A(p.R154Q)错义变异可能是该患儿的发病原因,因该变异在千人基因组数据库、人类基因突变数据库和Clinvar数据库及近期文献中未有该位点的相关报道,为未报道过的新变异,该新变异的检出丰富了NIPBL基因变异谱。经Sanger测序验证,该位点突变存在于患儿及患儿父亲。该突变位点位于5号染色体上NIPBL基因中,遗传方式为常染色体显性遗传,该患儿发病;携带同一基因的父亲有部分阳性体征,但临床特征评分<4分者不能诊断CdLS,推测患儿父亲为外显不全。常染色体显性遗传的家系中偶见携带致病基因突变个体不患病,表现为不完全外显,该个体称为顿挫型,顿挫型可不表现显性形状或部分表现,但可以把显性基因传递下去,使后代具有显性症状。在系谱中,由于顿挫型的存在常染色体显性遗传可以出现隔代遗传。如BOYLE等[7]2017年报道了1例CdLS患者的母亲及姨妈携带与患者相同的RAD21基因单碱基对缺失,但不符合CdLS的临床诊断标准,考虑与RAD21基因相关的外显不全有关。
CdLS是一种罕见的多系统遗传疾病,目前临床认可的与CdLS发病相关变异基因有NIPBL、SMC1A、SMC3、RAD21、HDAC8、BRD4、ANKRD11、AFF4、ARIDIB、EP300、KMT2A、NAA10、SETD5、SMARCB1、TAF6、ZMYND11、PHIP、MED13L、MAU2等[8-11]。不同的基因变异所导致的临床表现存在一定的差异,其中70%的患儿发病与NIPBL基因变异有关,但并不是所有NIPBL基因变异都能导致CdLS。NIPBL编码的Nipped-B-like(NIPBL)蛋白是Cohesin复合体的装载蛋白,鉴定为真菌和苍蝇姐妹染色单体内聚蛋白2(SCC2)的人类同源物,它与SCC2形成复合体,是黏着蛋白装载到染色体上所必需的,在染色质结构和转录调控中发挥重要作用[12-13]。在其他的发病相关变异基因中,如SMC1A、SMC3,已被证实与黏着蛋白有直接关系,而如ANKRD11等编码基因,则不直接相关。根据有无黏着蛋白相关基因突变,可将患儿分为经典型及非经典型,部分患儿可同时有经典型及非经典型相关基因突变。本例患儿基因突变位点位于NIPBL基因,考虑为经典型。
CdLS患儿除特殊面容、肢体畸形外,还可出现严重的智力障碍,包括交流能力下降、重复行为和自残行为以及多动,未来可能发展为自闭症。其中交流能力下降主要表现为口语的落后,多通过手势来表达[14-15]。因此,早期确诊并进行干预对提高患儿的语言行为能力、保持其心理健康具有重要意义。随着产前检查技术水平的提升,针对CdLS的产前诊断越来越多,胎儿宫内发育迟缓、特殊面容、多系统畸形(如心脏畸形、消化道畸形等)在产前胎儿超声中均可发现。对发现以上临床特征的胎儿,以及有CdLS家族史的高危人群,可对孕妇进行羊水穿刺、收集胎儿脱落的细胞行基因检测以明确诊断,实现优生优育。
目前针对CdLS尚无有效的治疗方法,主要采取对症支持治疗及康复训练。根据系统受累程度不同,针对患儿的随访、管理是不同的,发育迟缓的患儿应及早进行康复治疗干预,多脏器畸形如先天性心脏病等可进行针对性治疗及护理。CdLS患儿应根据病情定期随访,随访项目根据患儿年龄、疾病严重程度及累及系统制定。该例患儿在门诊康复治疗1个月后发育评估报告较前好转,但落后于正常同龄儿。研究显示,CdLS的发病机制与多种信号通路相关,其中包括经典的WNT通路。近期有研究显示,氯化锂可通过活化经典WNT通路,对CdLS模型有改善作用,提出锂作为CdLS的可能治疗策略[16],为临床治疗提供了一种新的选择。
综上所述,CdLS临床主要表现为智力发育落后、特殊面容、宫内和生后生长迟缓及多脏器畸形等,可结合基因结果进一步明确诊断。本例CdLS诊断明确,NIPBL基因c461G>A(p.R154Q)错义变异可能是其发病原因,该变异为新发基因突变,丰富了NIPBL基因变异谱。
