金属-有机框架材料在液相催化化学制氢中的研究进展
2022-01-24李淑蓉陈玉贞江海龙
李淑蓉,王 琳,陈玉贞,江海龙
(1.青岛大学化学化工学院,青岛 266071;2.中国科学技术大学化学系,微尺度物质科学国家研究中心,合肥 230026)
随着全球人口的日益增长和经济的快速发展,对能源的需求也急剧增加.但由于石油、煤炭等不可再生资源的逐渐枯竭及带来的环境污染问题日益突出,开发安全、清洁、可持续的新能源,或改善现有的能源储存和转换技术已成为必要的研究课题.氢能源作为一种全球公认的清洁燃料,具有来源广泛、能量密度高及常温下性质稳定等优点,且燃烧产物为水,绿色无污染[1].因此,氢能源的开发和利用可大大减少对化石燃料的高度依赖,对未来新型能源的可持续发展和解决环境污染问题具有重要参考意义.然而氢气(H2)的易燃性使其储存及运输面临诸多危险,大大限制了其实际应用.例如,氢燃料电池作为便携式电子设备和汽车的能量载体使用时,应具有尽可能高的能量含量和尽可能小的体积和质量,因此,开发安全高效的储氢方式逐渐成为人们关注的焦点[2].
氢气的储存方式主要有物理储存和化学储存两种.压缩气态氢、液态氢或通过高比表面积材料进行吸附等物理存储方法往往具有成本较高、安全性较差、质量大且不方便运输等特点[3].与物理储氢相比,以化学键形式存储氢的化学储氢方式更加安全、方便和高效,有望进行大规模推广使用[4].近年来,氨硼烷(NH3BH3,AB)、甲酸和水合肼等含氢量较高的化学储氢材料引起了学术界和工业界的广泛关注[5,6].甲酸中氢的质量分数为4.4%,具有对环境和生物无害的优点[7];水合肼中氢的质量分数高达8.0%,可以选择性地分解为氢和氮[8].氨硼烷具有高达19.6%的储氢质量分数及良好的储氢和放氢性能,并且无毒、稳定、对环境无污染,已然成为储氢材料的首选[9].这些储氢材料在催化剂条件下遇热分解或液相催化分解时,开始释放氢分子,并且水解产生的H2很容易从水中释放,方便收集利用.为了提高制氢效率、实现氢能源的大规模应用,需要合成高效、稳定的催化剂.金属纳米颗粒(MNPs)[10],尤其是贵金属催化剂,因其表面独特的电子结构和物理化学性质,往往具有较好的催化性能,也常被用来催化储氢材料产氢,并能显著提高制氢效率.但小尺寸的金属颗粒因具有的表面自由能较高,稳定性较差,且催化过程中易聚集,从而导致活性下降或循环性能变差[11].因此有必要寻找一种稳定、多孔、易制备的多孔材料来负载MNPs并防止其聚集.
金属-有机框架(MOFs,又称多孔配位聚合物或PCP)是一类新兴的多孔晶态材料,由金属节点(也称为二次建筑单元,或SBUs)和有机连接体配位构成,具有可调节和高度有序的孔结构[12~17].由于MOFs由于具有独特的结构特性而在气体储存和分离、传感、生物制药、能源和工业催化等领域表现出非常广泛的应用前景[18~30].利用MOFs分等级的多孔特性,将金属纳米粒子封装在框架孔洞来可阻止其在反应过程中发生团聚,提高催化活性,改善循环稳定性[31,32].近年来,以MOFs为前驱体,通过高温煅烧制备杂原子掺杂的多孔碳或多孔的金属/金属氧化物等多种碳基纳米复合催化剂备受关注.这种由MOFs模板法制备的碳基纳米材料不仅高度继承了MOFs的诸多优势,如规则的形貌、高比表面积、大的孔隙率等,还衍生出许多MOF不具备的优点,如高的热稳定性及具有催化活性位点的杂原子均匀掺杂等,极大地丰富了衍生材料的多功能性[33,34].本文综合评述了近年来MOFs、MOF基纳米复合材料以及MOF基衍生材料在催化氨硼烷、甲酸和水合肼等化学储氢材料水解制氢方面的研究进展(Scheme 1).同时对一锅串联反应—催化制氢和不饱和双键或硝基化学物的加氢反应也进行了简单概括,总结了MOF基纳米复合材料在液相催化化学制氢领域面临的机遇与挑战.表1列举了近年来MOF催化制氢方面的相关代表性文献[35~61].
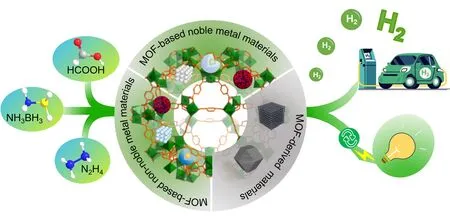
Scheme 1 Schematic hydrogen production from chemical hydrogen storage materials catalyzed by MOF⁃based composites
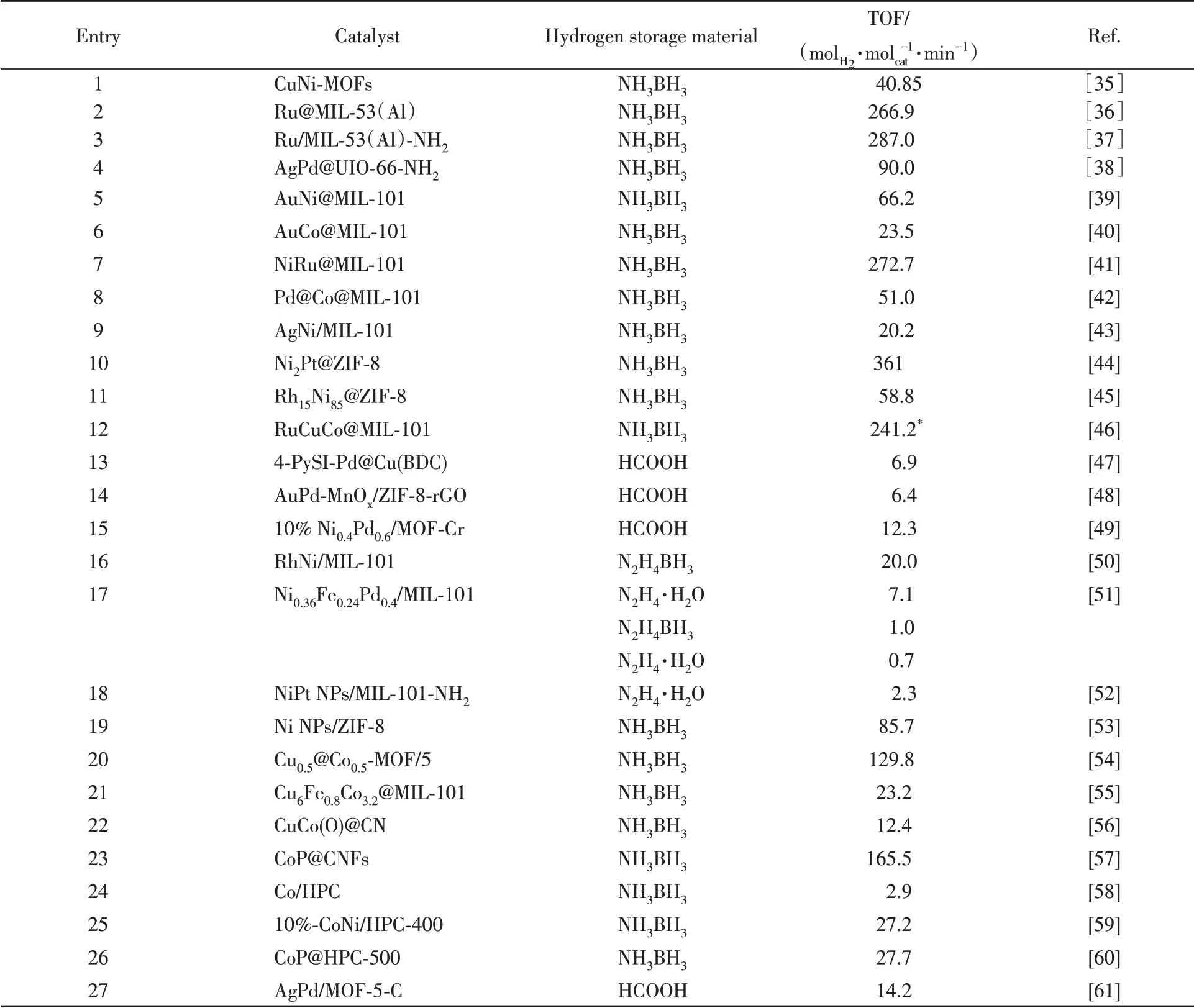
Table 1 Recent reports in catalytic hydrogen production by MOF-based composites
1 基于MOFs的催化化学制氢
由于MOFs材料具有多孔结构可调节,比表面积高及结构可灵活剪裁等特点,使其在能源开发领域具有广阔的应用前景.氨硼烷的高含氢量、低分子量以及高的溶液稳定性使其在过去几十年成为化学储氢的主要研究对象[9].氨硼烷可通过热解、水解以及醇解的方式释放氢气,其醇解方式与水解类似.本文主要总结了在MOF基催化剂存在下,氨硼烷的液相水解制氢方面的研究进展,并对氨硼烷高温条件下的热解制氢进行简单介绍.研究发现,将氨硼烷分子限制在MOFs孔洞中可有效改善热分解制氢效果,降低热分解温度,并抑制副产物生成[62].Wang等[62]探索了MOFs限域的AB(AB@MOF)热分解制氢温度与MOFs孔径大小间的联系.结果表明,氨硼烷颗粒大小受MOFs孔径限制,并且热分解温度与氨硼烷颗粒尺寸成正比,即AB@MOF中氨硼烷颗粒越小,其分解温度越低,此结果也与建立的氨硼烷分解热力学模型相吻合.
氨硼烷热分解制氢通常需要高温环境,且常伴有副产物的产生,通过MOFs的限域虽然可以降低其分解温度,但对反应设备和催化剂的稳定性要求较高.因此,长远来看,能够实现在温和条件下储氢材料的有效制氢是十分必要的.氨硼烷水解制氢可在室温下进行,是一种温和、绿色且安全的制氢方法,反应式可简单地表示为
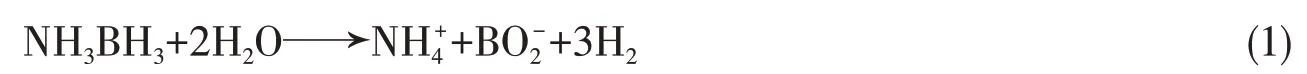
Zheng等[35]通过溶剂热法制备了分层多孔CuNi双金属MOF(CuNi-MOFs),该MOF不仅对催化氨硼烷水解制氢具有高活性,还具有良好的耐久性,且转化频率(TOF)可达40.85 molH2·molM-1·min-1,表观活化能为28.99 kJ/mol.但是目前极少有文献对单独的MOF催化NH3BH3水解进行报道,而以MOF基纳米复合材料与MOF衍生材料居多.
2 MOF基纳米复合材料的催化化学制氢
2.1 MOF基贵金属纳米复合材料
贵金属纳米粒子(Pt,Pd,Ag,Au,Ru,Rh等)由于其独特的物理化学性质,对各类化学反应都表现出优异的催化性能.通过MOFs的限域和稳定效应可有效减少金属纳米粒子在溶液中的聚集,提高其催化活性[63].此外,MOFs本身也具有一定的催化活性位点,可与金属纳米粒子的催化位点结合,实现协同催化的功效[64].
2.1.1 催化氨硼烷制氢目前,虽然多种催化剂被陆续研究出来用于提高氨硼烷水解制氢效率,但已报道的催化剂大部分集中于贵金属或贵金属掺杂的复合材料.例如,Muto等[65]利用超临界二氧化碳(scCO2)的辅助,在MIL-101(Cr)孔隙中引入H2PtCl6前驱体,随后在H2和N2氛围下还原得到Pt@MIL-101(Cr),并通过透射电子显微镜(TEM)证明了Pt纳米粒子(NPs)在MIL-101(Cr)中均匀分布.与传统方法合成的Pt@MIL-101(Cr)相比,通过scCO2辅助合成的Pt@MIL-101(Cr)对催化NH3BH3水解制氢具有更优异的催化活性.这是由于scCO2加速了H2PtCl6进入MIL-101(Cr)的中孔和微孔,提高了H2PtCl6在MIL-101(Cr)孔内的担载量,并且scCO2的高扩散和低黏度有利于Pt固定在MOFs的孔隙中.Zhou等[36]采用简单的液体浸渍方法首次在MIL-53(Cr)和MIL-53(Al)上成功沉积了超细Ru NPs,并使用该催化剂进行催化NH3BH3水解制氢.实验结果表明,2.65%Ru@MIL-53(Cr)和2.59%Ru@MIL-53(Al)表现出较高的催化活性,这源于Ru NPs在MIL-53上高度分散以及Ru NPs与MIL-53载体之间的协同效应.二者的TOF值分别为260.8和266.9 molH·2molR-u1·min-1,活化能分别为28.9和33.7 kJ/mol.基于以上研究,Zhou等[37]又通过原位浸渍还原法将Ru NPs固定在氨基功能化的MIL-53(Al)孔内,得到Ru/MIL-53(Al)-NH2催化剂,与Ru/MIL-53(Al)催化剂相比,其在常温下对NH3BH3水解制氢表现出更优的催化性能,TOF值高达287 molH·2molR-u1·min-1,活化能为30.5 kJ/mol.MIL-53(Al)-NH2具有更高的催化活性是由于氨基基团阻止了Ru NPs的团聚,促进了超小Ru NPs的形成与稳定.此外,Ru/MIL-53(Al)-NH2还具有良好的耐久性与可重用性,在5次循环的耐久实验和重复使用实验中,分别保持了其初始活性的72.4%和86.3%.
由于金属间存在协同催化作用,多金属合金或核壳结构的复合材料通常拥有比单金属催化剂更好的活性.Wang等[38]通过原位共还原法将双金属AgPd纳米粒子固定在UIO-66-NH2上,得到的双金属AgPd@UIO-66-NH2具有比单金属的Ag@UIO-66-NH2和Pd@UIO-66-NH2更好的催化NH3BH3水解制氢活性.通常,合金NPs的性能可以通过改变组成和原子的比例进行调节,在合成的不同Ag/Pd摩尔比的AgPd@UIO-66-NH2中,Ag1Pd4@UIO-66-NH2具有最好的催化活性,反应的TOF值达到90 molH2·
向贵金属纳米材料中引入过渡金属,形成贵金属-非贵金属双金属纳米材料,不仅可以减少贵金属的消耗,还能够提高催化活性.Xu等[39]采用双溶剂法(DSM),利用MOF内外部亲水性的不同以及毛细作用将Au3+和Ni2+前驱体引入MIL-101孔中,并首次结合液相浓度控制还原(CCR)策略控制Au3+和Ni2+前驱体还原过程中AuNi NPs的大小和位置(图1).他们发现,当采用强还原剂进行还原(OWR)时,超细AuNi NPs迅速被包裹在MIL-101的孔隙中,并且呈现均匀的三维分布.相反,如果选择温和还原(MR)方法,MNPs可能会在MOF表面发生严重的团聚.将得到的催化剂用于催化NH3BH3水解制氢,发现由于金属间的协同作用,双金属AuNi@MIL-101具有比单金属Au@MIL-101和Ni@MIL-101更高的活性,TOF值为66.2-1.此外,通过高浓度还原剂(0.6 mol/L NaBH4)还原得到的AuNi NPs具有更好的催化活性及稳定性,这是由于其颗粒尺寸小,提高了催化活性;MIL-101孔隙的限域作用阻止了MNPs的团聚,提高了稳定性.
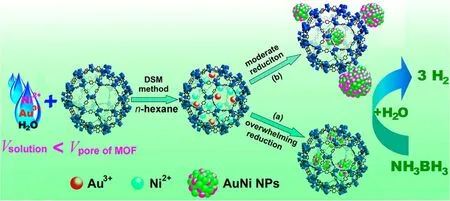
Fig.1 Schematic representation of immobilization of the AuNi nanoparticles by the MIL⁃101 matrix using the DSM combined with a liquid⁃phase CCR strategy[39]
Xu等[40]进一步通过DSM结合OWR方法成功地将超细AuCo NPs限域在MIL-101的孔隙中,其对催化NH3BH3水解制氢表现出优异的催化活性,TOF值为23.5-1,超过了单金属Au和Co.Xu等[41]进一步通过类似方法成功制备了MIL-101固定超细单分散NiRu NPs,与NiRu和Ni@Ru核壳NPs相比,NiRu@MIL-101催化剂对NH3BH3水解制氢反应表现出较高的活性和持久性,TOF值为
Jiang等[42]报道了一种由MOF稳定微小核壳Pd@Co NPs的新合成方法,其成功合成的关键在于将体积小于MOF孔体积的金属前驱体预先引入MOF孔中,然后利用Pd2+/Pd和Co2+/Co不同的氧化还原电势以及合适的还原剂,氨硼烷,将Pd2+和Co2+原位还原(图2).合成的Pd@Co@MIL-101不仅成本低,而且与单金属和合金相比,在NH3BH3的水解制氢反应中表现出协同增强的催化性能,TOF值为51 molH2·活化能为22 kJ/mol.此外,与Pd@Co负载在MOF外层的Pd@Co/MIL-101催化剂相比,Pd@Co@MIL-101具有更优越的催化活性和优异的循环稳定性,这可能是因为Pd@Co@MIL-101催化剂中Pd@Co NPs被限域和稳定在MOF孔内的,在反应过程中不容易发生颗粒迁移和团聚,提高了催化效率.
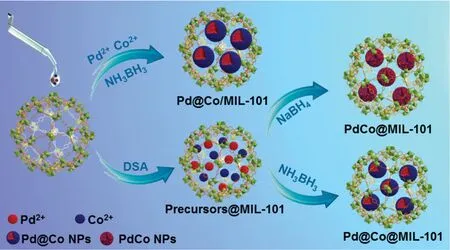
Fig.2 Synthesis of Pd@Co@MIL⁃101,Pd@Co/MIL⁃101,and PdCo@MIL⁃101 catalysts by different procedures and reducing agents[42]
为了降低贵金属含量,Jiang等[43]成功设计了一种新的简单的一步合成方法—贵金属种子诱导(NMSM)法,即利用微量的贵金属作为种子和引发剂,在温和的条件下还原非贵金属前驱体,原位生成MOF稳定的金属催化剂.在合成过程中,氨硼烷先将具有高还原势的贵金属离子还原,随后形成具有强还原性的M-H,进一步诱导非贵金属离子还原.成功制备的含有极低贵金属含量的AgNi/MIL-101催化剂(Ag/Ni摩尔比为1/200)经MOF稳定后,不仅成本降低,在室温条件下对NH3BH3的水解制氢反应也表现出良好的催化活性,TOF值为20.2
Astruc等[44]采用共还原法制备了一系列组分可调、无配体的近单分散双金属NiPt合金NPs,并将其嵌入ZIF-8中.由于NiPt之间存在强烈的协同作用,得到的NiPt@ZIF-8比单金属Ni@ZIF-8及Pt@ZIF-8具有更好的催化活性.对于催化NH3BH3水解制氢,Ni2Pt@ZIF-8表现出最好的性能,TOF值为361.此外,NaOH的存在可以促进H2的析出:在NaOH存在的条件下,Ni2Pt@ZIF-8催化NH3BH3水解制氢的TOF值可达到600
Luo等[45]采用简单的液体浸渍方法,成功地将高分散的平均直径为(1.1±0.2)nm的双金属RhNi纳米颗粒固定在ZIF-8上,在不同组成的催化剂中,Rh15Ni85@ZIF-8对NH3BH3水解的催化活性最高,TOF值为58.8
三金属组分催化剂具有更好的电子结构可调控性,其多个催化活性位点的协同应用及独特的性能吸引着人们的关注.Zhou等[46]首次成功合成了MIL-101负载的三金属RuCuCo纳米颗粒催化剂.由于强的三金属协同效应,RuCuCo@MIL-101对NH3BH3水解表现出很高的催化活性和稳定性,优于相应的单金属Ru和双金属CuCo,其催化NH3BH3水解反应的TOF值为241.2 molH2·molRu-1·min-1,活化能为48 kJ/mol.
2.1.2催化其它化学储氢材料制氢甲酸(HCOOH,FA)是一种稳定性高、无毒、方便且安全的储氢材料,可催化分解为H2和CO2,但同时也会产生有害气体CO,毒化催化剂的活性位点.

传统的甲酸选择性分解制氢是在有机金属配合物的均相催化剂催化下进行,通过加入添加剂(如甲酸钠或胺加合物等)来提高催化效率,但此方法容易造成催化剂与反应混合物的分离困难,限制了催化剂的规模化应用[66].近年来,利用MOF基贵金属纳米复合材料在温和条件下催化甲酸选择性制氢逐步受到大家的关注[47~49].
Rostamnia等[47]用一种独特的方法合成了OMS-Cu(BDC),并将其作为Pd-Schiff碱配合物的固体载体,将得到的4-PySI-Pd@Cu(BDC)用于甲酸的室温制氢反应,实验结果表明,4-PySI-Pd@Cu(BDC)对催化甲酸室温制氢有非常优异的催化活性,TOF值为412 h-1.Cu-MOF的多孔结构和Pd离子周围的Schiff碱基团对稳定Pd离子起着重要作用,Cu-MOF中开放的金属位点以及Pd金属与Schiff碱基团的协同效应对其催化性能起着关键的作用.
Zhu等[67]首次采用了一种简单的一锅合成法成功制备了新型核壳结构的纳米复合材料AgPd@MIL-100(Fe)(图3).首先,在140℃下,DMF在20 min内将AgNO3和Pd(NO3)2还原为AgPd NPs;随后,MIL-100(Fe)在PVP修饰的AgPd NPs表面自发生长,生成均匀的核壳AgPd@MIL-100(Fe).在上述反应中,DMF既作为H3BTC的溶剂,决定MIL-100(Fe)晶体的生成,又作为还原剂,将金属离子还原为AgPd NPs.这种新型核壳结构催化剂AgPd@MIL-100(Fe)在室温、不添加任何试剂的情况下就可高效催化甲酸分解制氢.与单独的AgPd NPs相比,双金属核壳结构催化剂AgPd@MIL-100(Fe)协同提高了甲酸在水溶液中的制氢催化活性,而且,经过多轮反应后仍能保持高催化活性.

Fig.3 Schematic representation of synthesis and application of AgPd@MIL⁃100(Fe)core⁃shell NPs for FA decomposition at 298 K[67]
Jiang等[48]以ZIF-8和还原氧化石墨烯(rGO)为双载体,采用简单的湿化学方法成功制备了一种AuPd-MnOx/ZIF-8-rGO纳米催化剂.该催化剂对甲酸水溶液制氢具有较高的活性,298 K时TOF值达到382.1 molH2·molcat-1·h-1.其良好的催化活性源于AuPd-MnOx/ZIF-8-rGO中Pd电子结构的改变、超细AuPd-MnOx在双载体ZIF-8-rGO中均匀分散、以及AuPd-MnOx与ZIF-8-rGO之间强的金属-载体相互作用.
通过向贵金属中掺入非贵金属形成双金属NPs也可提高甲酸分解制氢的催化活性.Chang等[49]采用过度浸渍-低温还原的方法合成了以MOF为载体的钯基催化剂,用于催化甲酸分解制氢.在所合成的催化剂中,10%Pd/MOF-Cr表现出显著的性能,在323 K下TOF值为537.8 h-1,进一步合成的双金属10%Ni0.4Pd0.6/MOF-Cr具有更优的催化性能,323 K下TOF值可达737.9 h-1.实验结果进一步表明,双金属间的协同作用可以提高催化甲酸水解制氢活性.
除氨硼烷和甲酸外,水合肼(N2H4·H2O)和肼硼烷(N2H4BH3)也是高含氢量化学储氢材料,其分解方式如下:
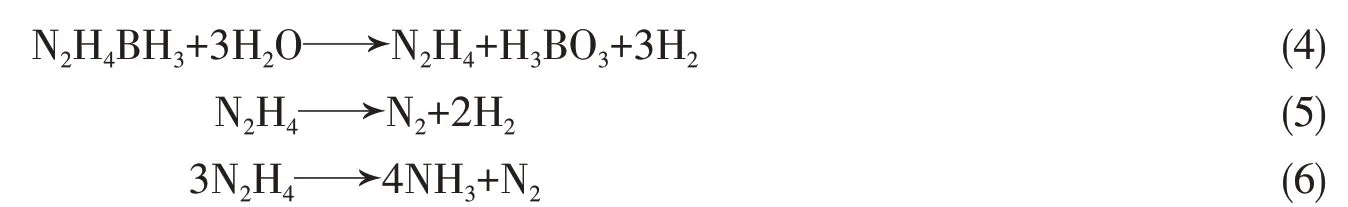
为了实现N2H4·H2O与N2H4BH3100%的氢利用效率,应避免N2H4通过式(6)分解产生NH3和不需要的产物.迄今,大多数报道的催化剂仅对第一步水解[式(4)]有效,且N2H4的选择性分解仍然高度依赖于贵金属基催化剂.
Lu等[68]通过一种控制还原速率的策略,将均匀细小的NiPt合金纳米团簇封装在MOF开放的多孔通道内,所合成的Ni0.9Pt0.1/MIL-101核壳复合材料具有极低的Pt含量,在碱性N2H4BH3和N2H4·H2O溶液中具有极高的活性粒径和耐久性,且能够实现100%的氢选择性.NiPt/MIL-101良好的催化活性是由于NiPt NPs粒径较小、NiPt NPs与MOF之间具有较强的协同效应以及开放的MOF孔结构便于自由传质.Lu等[50]采用相似的方法制备了MIL-101稳定的小尺寸RhNi合金NPs,所得到的RhNi/MIL-101催化剂具有高的活性和耐久性.其中优化后的Rh0.8Ni0.2/MIL-101催化剂对催化水溶液中的N2H4BH3和N2H4·H2O制氢具有优异的性能,转化率和氢选择性均为100%,TOF值分别达到1200和428.6 molH2·,高于Rh基催化剂.Lu等[51]还通过简单的浸渍法制备了MIL-101稳定的三金属NiFePd纳米粒子,与单独的Ni0.36Fe0.24Pd0.4NPs、MIL-101负载的单金属和双金属催化剂相比,在323 K下,Ni0.36Fe0.24Pd0.4/MIL-101催化剂对N2H4BH3和N2H4·H2O水解制氢表现出更高的催化性能,TOF值分别为60和40.8 h-1.
在MOF上修饰官能团可能会对催化反应产生一定的影响,改善催化活性.Su等[52]合成了MOF稳定的双金属NiPt和NiIr合金纳米颗粒,并探究了MOF中嵌入的一系列官能团对水合肼完全转化为氢气的催化性能.与无官能团修饰和受电子基团—NO2和—SO3H功能化的MIL-101稳定NiPt NPs相比,供电子基团—NH2功能化的NiPt NPs/MIL-101表现出更高的活性,TOF值在293 K时为137 h-1.这可能是由于活性NiPt NPs在功能化MOF中的电子态不同以及水合肼分子与MOF载体中官能团的相互作用强度不同所致,此外,载体的组成和孔结构对催化剂的催化活性也有显著影响.
Xu等[69]首次采用一步还原法制备了一系列高效的RhNiP@MOF-74复合催化剂.由于Rh,Ni,P之间的协同电子效应以及RhNiP和MOF-74之间的电子转移现象,催化剂在碱性溶液(2 mol/L NaOH)中对水合肼制氢表现出优秀的催化性能.其中,Rh47Ni18P35@MOF-74催化水合肼制氢的TOF值达到715.4
Zhao等[70]采用简单、安全的化学浸渍法制备了一种负载在UiO-66上的NiFePd催化剂,得到的NiFePd/UiO-66具有较高的比表面积,并且具有高效的在碱性溶液中催化水合肼制氢性能.其中Ni0.25Fe0.25Pd0.5/UiO-66催化剂在碱性水溶液中表现出最佳的氢选择性,TOF值在80℃下可达662.4 h-1.
2.2 MOF基非贵金属纳米复合材料
虽然贵金属催化剂对催化化学储氢材料制氢具有优异的催化活性,但由于其成本较高、储量有限,大大限制了其大规模应用,与贵金属相比,非贵金属[71]价格低廉、储量丰富,因此发展高效、稳定、低成本的非贵金属催化剂对化学储氢材料制氢的实际应用具有重大意义.
非贵金属催化剂催化NH3BH3水解制氢已被陆续报道[72],利用廉价且储量丰富的第四周期过渡金属(Cu,Co,Ni和Fe等)在温和条件下催化NH3BH3水解制氢的实际效率和可持续性仍然极具挑战性[73],这主要因为过渡金属离子不易被还原且小尺寸颗粒容易团聚.MOF的多孔结构可以限制和稳定非贵金属纳米颗粒,从而控制金属纳米颗粒的成核和生长,阻止其聚集并提高其稳定性.Astruc等[53]以ZIF-8为载体,合成了具有显著催化效率的过渡金属催化剂,MNPs/ZIF-8,并对其催化NH3BH3水解制氢效果进行了比较,发现Ni NPs/ZIF-8催化效果最好,室温下TOF值达到85.7.基于动力学同位素效应研究,通过氧化加成H2O中的一个O—H键来裂解是决定反应速率的步骤.他们在这些机理研究的启发下,进一步提出了一种有趣的、高效的制氢“开-关”控制方法,即通过向反应介质中加入等摩尔量的HCl和NaOH水溶液来实现对氢气生成的“开-关”控制(图4).
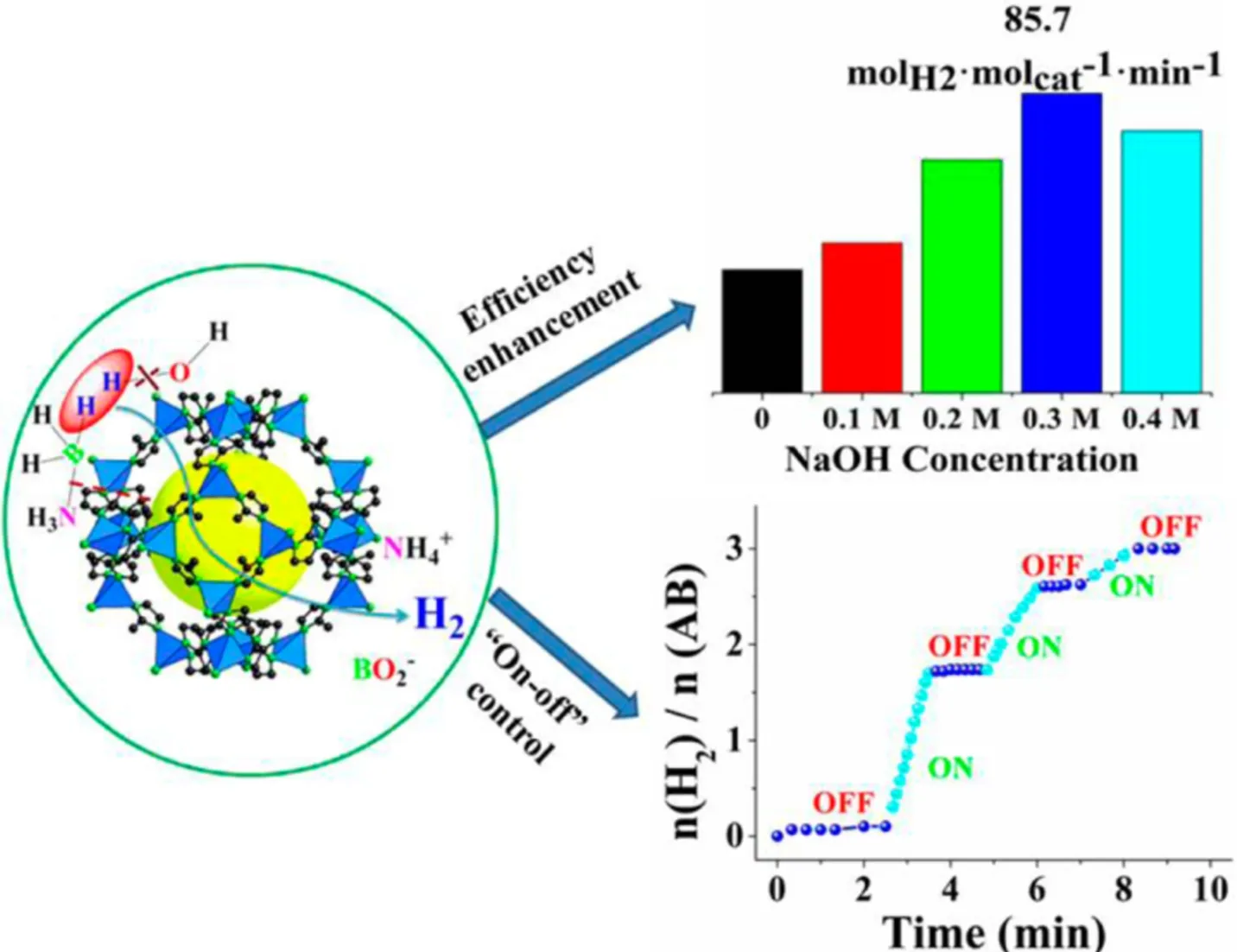
Fig.4“On⁃off”control of H 2 production in the NH 3BH 3 hydrolysis in water[53]
一些等离子体催化剂(Ag,MoO3-x和Pd/MoO3)可以显著改善NH3BH3水解的催化活性,这是因为光激发电荷的迁移可以通过富电子NH3部分或缺电子BH3部分促进氨硼烷的活化,并且在光催化反应过程中产生的活性物种进一步提高了催化活性[74].Yamashita等[74]首次证明了在可见光照射下,负载在MIL-101上的非贵金属纳米颗粒可以显著提高氨硼烷制氢的催化活性,且在可见光照射下的催化活性明显高于无光照射时的催化活性.此外,这些催化剂也表现出较高的稳定性,在可见光照射下产生的超氧阴离子和羟基自由基对实现高催化活性起着至关重要的作用.Cu/MIL-101,Co/MIL-101和Ni/MIL-101对于催化氨硼烷制氢的TOF分别为1693,1571和3238 h-1.
MOF并不局限于作为承载金属NPs的载体,它们暴露的金属节点也可以作为反应活性位点,并且与双金属的活性中心的协同效应可以显著提高反应的活性.Li等[54]通过溶剂热和适度活化过程,使Cu NPs在钴金属有机框架(Co-MOF)中原位生长,得到的Cu@Co-MOF催化剂具有丰富的双位点.优化后的Cu0.5@Co0.5-MOF/5对NH3BH3水解制氢反应的TOF值为129.8,反应活化能为26.5 kJ/mol,经过10轮循环稳定性保持不变.基于密度泛函理论(DFT)计算,这种显著提高的活性源于Cu NPs和Co-MOF之间独特的界面电子结构,其中O-Co2+位点可以有效激活水分子,Cu NPs位点可以成功激活氨硼烷分子,Cu-Co2+双活性位点的协同作用使氨硼烷水解更容易进行.
由于金属间良好的协同作用,合成多金属非贵金属纳米颗粒是提高催化活性的行之有效的方法.Xu等[75]通过DSM结合OWR方法成功将双过渡金属CuCo NPs引入到MIL-101的孔隙中.与单金属相比,双金属CuCo NPs在室温下对NH3BH3水解制氢具有更高的催化活性,铜钴物种间的协同效应对催化NH3BH3水解性能的提高起着重要作用.
Gu等[76]提出了4种简单的制备方法用于合成非晶态或晶态的多孔MIL-101负载Co NPs催化剂.其中,超声辅助原位合成的无定形小尺寸Co NPs催化剂具有最好的活性.此方法也成功地应用于双金属体系的合成,所得MIL-101负载的非晶CuCo,FeCo和NiCo NPs催化NH3BH3制氢的TOF值分别为51.7,50.8和44.3 molH2·molcat-1·min-1,超过很多已知的单质金属,甚至贵金属催化剂.
Li等[55]通过原位浸渍还原法在MIL-101孔隙中引入了不同摩尔比的CuFeCo三金属纳米粒子,与双金属催化剂相比,Cu6Fe0.8Co3.2@MIL-101对NH3BH3水解制氢的催化性能更好,TOF值为23.2 molH2·,反应活化能为37.1 kJ/mol,并且在循环7次后仍保持稳定.
2.3 MOF基衍生材料
碳基纳米材料在化工生产、能源和环境中被广泛用作催化剂或催化剂载体,近年来,MOF因其高比表面积、丰富的金属/有机配体种类、大的孔隙体积及结构可调性等优点成为制备各种碳基纳米材料的优良模板和前驱体[77,78].将MOF在高温条件下煅烧转化为包覆金属纳米颗粒的多孔碳基复合材料,得到的多孔碳材料在反应过程中不易团聚,有利于反应底物和产物的有效运输.另外获得的催化剂比MOFs本身更加稳定,方便多次循环利用.近年来,MOF基衍生材料在催化领域以及在合理负载活性位点和杂原子功能化方面表现出很大潜力[77].
2.3.1催化氨硼烷制氢MOF基衍生材料的合成及应用虽然已有大量文献报道,但大多集中在电催化应用领域,在多相催化方面的报道相对较少,对催化NH3BH3水解制氢的报道更少.Xu等[79]研究了一种原位包覆高分散超细Ru纳米粒子的方法(图5),合成的Ru@HKUST-1复合材料在惰性气氛下热解生成的Cu/Ru@C对NH3BH3水解制氢具有非常高的催化活性和良好的耐久性.在Ru@HKUST-1复合材料的热解过程中,Cu/Ru纳米颗粒周围形成的石墨化碳有效抑制了纳米颗粒的团聚,使微小的纳米颗粒得以在碳基体中高度分散并稳定存在.
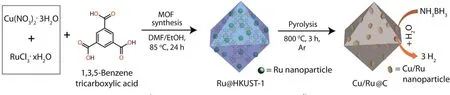
Fig.5 Schematic illustration of the synthesis of Ru@HKUST and Cu/Ru@C composites for AB hydrolysis[79]
Liu等[80]以Co-MOF和间苯二酚-甲醛树脂为前驱体合成了Co@NCS,随后又通过可控氧化方法将其氧化为Co-CoOx@NCS-n.由于Co和CoOx之间的协同效应,Co-CoOx@NCS-II在298 K下催化NH3BH3产氢的速率为5562 mL·min-1·gCo-1.
沸石咪唑类骨架材料(ZIF)是一种常用的制备氮掺杂多孔碳材料的前驱体.Kegnæs等[81]利用ZIF-67作为牺牲前驱体,制备了氮掺杂的多孔碳稳定的Co纳米粒子,并对其催化NH3BH3水解制氢性能进行了测试.结果表明,纳米Co颗粒的尺寸和碳载体的结构特征对催化活性有很大的影响.此外,在催化剂前驱体ZIF-67/8中添加Zn也会对催化剂性能产生影响,当Co/Zn摩尔比为1时,900℃下碳化蒸发除锌后,得到的催化剂活性最高.在室温下,该催化剂催化NH3BH3水解制氢的TOF值为7.6 molH2·molCo-1·min-1,表观活化能为44.9 kJ/mol.此外,在0.1 mol/L NaOH溶液中,TOF值还可进一步增加到12.7 min-1.
Wang等[56]通过热还原Cu离子掺杂的ZIF-67(CuCo-ZIF)前驱体合成了在碳-氮框架(CN)上分散良好的CuCo和CoO纳米粒子[CuCo(O)].所制备的CuCo(O)@CN保持了ZIF-67的多面体形貌、良好的纳米粒子空间分布和较多的活性位点.CuCo(O)@CN作为NH3BH3水解制氢催化剂时表现出优异的催化活性,TOF值为12.4,相应的活化能为33.8 kJ/mol.其优异的催化性能归因于Co和Cu之间的协同作用以及含氮量丰富的碳骨架.
过渡金属磷化物对催化NH3BH3水解具有很好的活性.以MOF为牺牲模板合成金属磷化物可有效减少磷化物合成过程中金属纳米粒子的聚集.Chen等[57]以双金属Co/Zn-ZIF为模板,成功合成了三维多孔的CoP碳基纳米框架(CoP@CNFs),并用作催化NH3BH3水解制氢的催化剂.MOF前驱体中的Zn在热解过程中起着分离Co离子和维持骨架的双重关键作用(图6).结果表明,多孔CoP@CNF具有较大的比表面积、分级的多孔结构、暴露的活性位点和亲水性通道,这些都有利于提高催化NH3BH3水解制氢的活性,其催化NH3BH3制氢的TOF值为165.5 molH·2molCo-1·min-1.
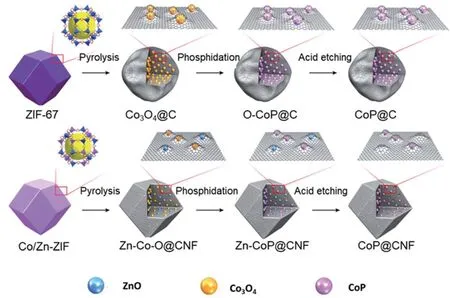
Fig.6 Schematic illustration of the synthetic procedure for the porous CoP@CNF[57]
Xu等[82]首先通过两步热处理将ZIF-67转化为含氧多孔碳材料(OPC-300),随后通过简单的一部化学还原法在OPC-300上负载NiCoP,得到NiCoP/OPC-300.其中Ni0.66Co0.19P0.15/OPC-300纳米催化剂在298 K、碱性条件下(0.4 mol/L NaOH)对NH3BH3水解制氢表现出最佳的催化活性,TOF值高达95.24 molH2·molcat-1·min-1.Xu等[83]又继续探究均匀分散的CoP纳米颗粒的合成方法,即通过将Co-MOF-74简单磷化得到CoP@CNR.他们发现热解温度对催化活性有一定的影响,CoP@CNR(400)对碱性条件下NH3BH3水解制氢具有较好的活性和稳定性,氢释放速率为10014 mL·min-1·g-1,第5次循环时仍能保持初始活性的92.7%,表现出了良好的稳定性.
Chang等[58]采用选择性锌蒸发分离策略,以双金属Co/Zn-MOF-74为牺牲模板,合成了一系列含有多级孔的碳骨架负载的Co NPs.前驱体中Zn的加入有效抑制了Co NPs在热解过程中的团聚.研究结果表明,不同Co/Zn的摩尔比和热解温度对催化NH3BH3水解制氢性能有着关键的影响,其中在900℃下热解的30%Co/HPC催化剂的催化活性最高,TOF值为176.5 h-1.Chang等[59]进一步通过原位碳化还原方法,采用双金属的CoNi-MOF-74作为牺牲模板,成功合成了高度分散在多孔碳上的超细CoNi纳米合金.精细的核壳结构能显著稳定CoNi NPs,抑制其在催化NH3BH3水解制氢过程中的聚集和流失,同时多孔壳层也有助于反应底物的快速传质.研究发现,通过改变MOF-74前驱体的Co/Ni比例或热解温度,可以有效地调节复合催化剂的催化性能.在所制得的复合材料中,10%-CoNi/HPC-400具有最佳的催化活性,其催化NH3BH3水解制氢的TOF值达到27.22 min-1.Chang等[60]又进一步考察了过渡金属磷化物催化NH3BH3水解制氢的效果.他们以Co-MOF-74为牺牲模板,采用分步煅烧和磷化法制备了一系列拥有多级孔的碳材料,并成功负载非贵金属CoP NPs,将所得CoP@HPC-T复合材料用于催化NH3BH3水解制氢的研究,结果表明,500℃下煅烧得的CoP@HPC-500的催化性能最好,其催化NH3BH3水解制氢的TOF值为27.7 min-1,活化能为42.55 kJ/mol.
2.3.2催化其它化学制氢材料制氢除了在NH3BH3水解制氢中应用,MOF基衍生材料在其它储氢材料制氢的研究中也有报道.Wang等[61]首次制备了一种高效的双金属协同催化剂—金属-有机框架多孔碳负载AgPd双金属(AgPd/MOF-5-C).在室温条件下,该催化剂表现出较高的甲酸制氢催化活性及100%的H2选择性,TOF值可达854 h-1.
负载型金属单原子催化剂(SACs)因具有更高的原子利用率和TOF值而备受关注.Beller等[84]利用MOF的高温热解合成了具有孤立钴单原子的衍生Co-N-C催化剂,并用于催化甲酸制氢.与采用类似方法合成的纳米颗粒相比,原子分散的Co-N-C催化剂表现出更高的活性、更好的耐酸性和长期稳定性.
3 化学制氢-有机反应的串联催化探究
由于氢气在多数溶剂中的分散性较差,而烯烃、炔烃和硝基化合物等有机物的加氢通常需要用氢气作还原剂,因此氢气分子与反应物充分接触是提高反应效率的重要条件.研究发现,储氢材料水解原位产出的氢气可高度分散在溶剂中,并能保持一定的时间,因此通过将储氢材料的原位制氢与有机化合物加氢还原串联起来是一个良好的研究方案,在整个反应体系中,原位生成的H2能够迅速与反应底物接触并将其还原,大大提高了反应效率[85].
Feng等[86]通过一步碳化NH2-MIL-125制备了N掺杂的多孔碳(N-MOF-C),随后以NaBH4为还原剂,采用共还原法将Pd和Ag纳米粒子固定在N-MOF-C上.结果表明,Pd9Ag1-N-MOF-C催化剂对催化甲酸制氢及硝基芳烃加氢串联反应具有良好的催化活性和选择性,该催化剂的高催化活性主要归功于多孔载体与高度分散的Pd和Ag金属纳米颗粒之间的协同作用.
在目前报道的MOFs基复合材料催化一锅多步串联反应,即化学储氢材料制氢和化合物加氢的反应中,大多选择含氢量高的氨硼烷为氢来源,而对甲酸及其它化学储氢材料的报道较少.Jiang等[87]通过双溶剂法将钯前驱体引入介孔MOF中,随后通过NH3BH3原位还原,得到MIL-101限域的Pd NPs(约3 nm).得到的Pd@MIL-101在催化NH3BH3水解制氢和硝基化合物还原的串联反应中表现出良好的催化性能,特别是硝基苯加氢反应的TOF值高达97 molNitrobenzene·molPd-1·min-1,催化活性超过了以往报道的钯催化剂,此外,这也是金属NPs@MOFs首次以单金属NPs为唯一活性位点催化串联反应的报道.Jiang等[88]进一步将成本较低的CuNi过渡金属纳米粒子限制在MIL-101孔内,得到CuNi@MIL-101,其在温和条件下对催化NH3BH3制氢以及硝基芳烃的加氢串联反应表现出良好的循环性,这是首次报道MOF稳定的非贵金属NPs催化剂用于串联反应.
Chen等[89]成功合成了MOF稳定的小尺寸Cu@Co@Ni纳米颗粒(3.3 nm).由于三金属之间优异的协同催化作用,在室温条件下,该催化剂可在5 min内通过氨硼烷分解得到的原位氢将一系列硝基苯衍生物还原为相应的氨基化合物.与常规方法相比,该方法可将还原速率提升近20倍,且无需在高压氢气条件下进行,更加安全.此外,Cu@Co@Ni/MOF对催化CO氧化反应也表现出很好的催化性能,当温度升到180℃时CO转化率达到了100%,活性超过了大部分报道的贵金属催化剂.随后,本课题组[90]设计了一种简单的刻蚀合成策略,即通过调控配体的浓度和金属氧化物刻蚀的时间,成功实现了MOF在八面体形Cu2O纳米晶体(NCs)表面上的定向生长,获得同样具有八面体形貌的Cu2O@HKUST-1复合材料[图7(A)],随后用NH3BH3原位还原得到Cu@HKUST-1.这一催化剂有效结合了Cu NCs的等离子体光热(SPR)效应和氢化活性及MOF的Lewis酸性,实现了对级联反应—NH3BH3放氢和硝基化合物加氢的协同催化[图7(B)].此外,由于HKUST-1壳层的保护作用,复合材料的结晶度、形貌以及催化活性即使经过5次循环也能保持良好.据我们所知,这是首次尝试将金属纳米晶的SPR效应与MOFs的Lewis酸性进行结合,用于对单锅串联反应的协同催化.
4 总结与展望
由于氢能具有来源广泛、绿色无污染、能量密度高等优点,其开发和应用备受关注.MOFs基纳米复合材料在催化化学制氢方面具有独特的优势,虽然MOF基贵金属纳米复合材料对催化氨硼烷、甲酸、水合肼等化学储氢材料制氢展示出优异的催化性能,但由于贵金属价格昂贵且储量较低,限制了其在催化制氢方面的大规模应用.由于非贵金属价格低廉且储量丰富,MOF基非贵金属材料及MOF衍生的非贵金属@多孔碳材料在催化化学储氢材料制氢领域具有广泛的应用前景.不过,目前这方面的研究仍面临诸多挑战,如MOF基非贵金属材料的研究大多集中在催化氨硼烷水解制氢方面,对其它储氢材料的研究较少;催化材料的合成繁琐,成本高,催化效率有限,尚难以进行实际应用.为了实现氢能源安全高效的大规模开发和应用,需要深入探索MOF基非贵金属材料在多种化学储氢材料制氢方面的应用,尤其是将制氢反应与其它类型的反应相结合,实现多功能催化剂的高度利用和化学反应的串联协同.相信不久的将来,通过科学家们的不断努力和探索,氢能源的制备、运输及储存方案将会不断完善.这种清洁高效的新能源也将突破瓶颈逐渐走向生活,走向社会,成为人类经济发展的重要新型能源,而MOF基纳米复合材料也必将在氢能源的开发和利用中发挥重要作用.
