Diversity of Sodium Transporter HKT1;5 in Genus Oryza
2022-01-20ShaliniPulipatiSujiSomasundaramNitikaRanaKavithaKumaresanMohamedShafiPeterCivGothandapaniSellamuthuDeepaJaganathanPrasannaVenkatesanRamaraviPunithaKalaimaniRajuShrikantMantriSowdhaminiAjayParidaGayatriVenkataraman
Shalini Pulipati, Suji Somasundaram, Nitika Rana, Kavitha Kumaresan, Mohamed Shafi, Peter Civáň, Gothandapani Sellamuthu, 6, Deepa Jaganathan, Prasanna Venkatesan Ramaravi, S. Punitha, Kalaimani Raju, Shrikant S. Mantri, R. Sowdhamini, Ajay Parida, Gayatri Venkataraman
Research Paper
Diversity of Sodium Transporter HKT1;5 in Genus
Shalini Pulipati1, Suji Somasundaram1, Nitika Rana2, Kavitha Kumaresan3, Mohamed Shafi4, Peter Civáň5, Gothandapani Sellamuthu1, 6, Deepa Jaganathan7, Prasanna Venkatesan Ramaravi8, S. Punitha1, Kalaimani Raju1, Shrikant S. Mantri2, R. Sowdhamini4, Ajay Parida9, Gayatri Venkataraman1
(M. S. Swaminathan Research Foundation, Chennai 600113, India; National Agri-Food Biotechnology Institute, Punjab 140306, India; Krishi Vigyan Kendra, Kanyakumari 629901, India; National Centre for Biological Sciences / Tata Institute of Fundamental Research, Bangalore 560065, India; National Research Institute for Agriculture, Food and Environment University, Clermont- Auvergne, Clermont-Ferrand 63000, France; Forest Molecular Entomology Laboratory, Faculty of Forestry and Wood Sciences, Czech University of Life Sciences, Prague, Praha 16500, Czech Republic; Tamil Nadu Agricultural University, Coimbatore 641003, India; International Business Machines India, Chennai 600125, India; Institute of Life Sciences, Bhubaneswar 751023, India)
Asian cultivated rice shows allelic variation in sodium transporter, OsHKT1;5, correlating with shoot sodium exclusion (salinity tolerance). These changes map to intra/extracellularly-oriented loops that occur between four transmembrane-P loop-transmembrane (MPM) motifs in OsHKT1;5. HKT1;5 sequences from more recently evolvedspecies (/complex species) contain two expansions that involve two intracellularly oriented loops/helical regions between MPM domains, potentially governing transport characteristics, while more ancestral HKT1;5 sequenceshave shorter intracellular loops.We compared homology models for homoeologous OcHKT1;5-KandOcHKT1;5L from halophyticto identify complementary amino acid residues in OcHKT1;5-L that potentially enhance affinity for Na+. Using haplotyping, we showed that Asian cultivated rice accessions only have a fraction of HKT1;5 diversity available in progenitor wild rice species (and). Progenitor HKT1;5 haplotypes can thus be used as novel potential donors for enhancing cultivated rice salinity tolerance. Within Asian rice accessions, 10 non-synonymous HKT1;5 haplotypic groups occur. More HKT1;5 haplotypic diversities occur in cultivatedgene pool compared to. Predominant Haplotypes 2 and 10 occur in mutually exclusiveandgroups, corresponding to haplotypes insalt-sensitive and salt-tolerant landraces, respectively. This distinct haplotype partitioning may have originated in separate ancestral gene pools ofand, or from different haplotypes selected during domestication.Predominance of specific;haplotypes within the 3 000 rice dataset may relate to eco-physiological fitness in specific geo-climatic and/or edaphic contexts.
HKT1;5 diversity; single nucleotide polymorphism; haplotype; bacterial artificial chromosome; salinity tolerance; sodium transporter;species
Rice is a staple crop grown by millions of small- holder farmers. It shows enormous diversities and gene banks worldwide contain extensive seed collections, covering genetic diversity present in farmers’ cultivars, landraces andspecies (Menguer et al, 2017). Sustaining rice production depends heavily on the intelligent use of rice diversity in the context of changing environment and expansion of arable land into new areas. The genusconsists of 27 species, encompassing nearly 15 million years of evolution, consisting of 11 genome types, 6 of which are diploid (n = 12; AA, BB, CC, EE, FF and GG) and 5 are polyploid (n = 24; BBCC, CCDD, HHJJ, HHKK and KKLL) (Stein et al, 2018). Of these, both Asian and African cultivated rice (and, respectively) belong to the AA group. As wild relatives of rice show adaptation to defined bio-geographic ranges and can tolerate many biotic and abiotic stresses, they constitute an important reservoir for crop improvement. The InternationalMap Alignment Project, a wild rice genomic resource (Ammiraju et al, 2006; Stein et al, 2018) as well as the cultivated rice‘3K-rice genomes’ (3K-RG) dataset (The 3 000 rice genomes project, 2014; Wang W S et al, 2018) aim to catalogue natural variations that exist in cultivated and wild rice and utilize the information to identify genes and genomic regions that can be used to drive the next generation of crops in the era of climate change.
Globally, 45 Mhm2of irrigated and 32 Mhm2of rain-fed agricultural lands are affected by salinity (http://www.fao.org/soils-portal/en/). Cultivated rice,, is highly sensitive to salinity at both seedling and reproductive stages (Moradi et al, 2003). Salinity tolerance in many plant species, including rice, is inversely related to the extent of Na+accumulation in the shoots (Kumar et al, 2015; Hoang et al, 2016). High-affinity K+transporter (HKT) gene family members play a central role in controlling Na+accumulation and thus, in conferring salinity tolerance (Ren et al, 2005; Munns et al, 2012; Byrt et al, 2014; Yang et al, 2014; Jiang et al, 2018). HKT transporters are classified into two sub-types based on gene organization, protein structure and ion specificity as: sodium uniporters (Type I) and sodium/potassium symporters (Type II). Type I and II HKTs show characteristic differences in the selectivity filter (SF) conferring ion specificity: the amino acid in the first pore domain (PD) of the HKT protein is the main distinguishing feature (Platten et al, 2006). Members of Type I show a serine residue in the SF, while the remaining three amino acid residues in the SF are glycine (S-G-G-G motif). Type II HKTs show a ‘G’ at all positions in the SF (G-G-G-G motif). However, exceptions to this rule have been reported, implicating additional residues in conferring cation specificity (Ali et al, 2016).
While only one HKT geneoccurs in(Rus et al, 2001, 2004; An et al, 2017), dicot as well as monocot species show a multiplicity of HKT genes (Asins et al, 2013; Waters et al, 2013; Almeida et al, 2014; Véryet al, 2014). Natural variations in(allelic differences related to amino acid substitutions and/or expression differences) have been associated with conferring salinity tolerance in both.and cereal crops (among cereal crops, specifically Type I) (Munns et al, 2012; Busoms et al, 2018; Jiang et al, 2018). HKT1;5 is a plasma membrane transporter that retrieves Na+from root xylem vessels (loaded/transferred to adjacent parenchyma cells), reducing xylem sap Na+loading and minimizing Na+transport to shoot. Introgression of() frominto salt sensitive durum wheat (lacking) confers significant reduction in leaf Na+concentration and enhances grain yield on saline soils (Munns et al, 2012). Thelocus (harbouring) in bread wheat is associated with salinity tolerance, and RNAi silencing ofin transgenic bread wheat leads to leaf Na+accumulation (Byrt et al, 2014). In rice, allelic variation inat thelocus is associated with salinity tolerance, and four amino acid changes in the OsHKT1;5 sequence confers differential Na+transportcharacteristics (Ren et al, 2005; Thomson et al, 2010). Variations in;5 expression levels in the roots of rice landraces also directly correlate with salinity tolerance (Cotsaftis et al, 2012). In maize, two SNPs in the coding region ofare significantly associated with variations in salt tolerance (Jiang et al, 2018). More recently in barley, natural variation inexpression is associated with sodium exclusion from shoots (van Bezouw et al, 2019), and functionally compromised alleles of(due to mis-localization and/or altered Na+transport activity)increase shoot and grain Na+contents (Houston et al, 2020). Thus, allelic variation inhas the potential to affect shoot Na+content and hence, plant fitness under salinity.
diversity has been examined inaccessions in relation to salinity tolerance through microarray, allele mining,Ecotype Targeting Induced Local Lesions IN Genomes (EcoTILLING) and genome-wide association study (GWAS) approaches (Cotsaftis et al, 2012; Negrão et al, 2013; Platten et al, 2013; Yang et al, 2018), and in select wild rice AA genome species, through PCR sequencing based approaches (Mishra et al, 2016). Among other wild rice species,has been examined in detail only in halophytic(Somasundaram et al, 2020). The study surprisingly showed that the OcHKT1;5-K homolog encodes a low affinity Na+transporter whileis a species growing in saline habitats. Crucial amino acid changes conferring low affinity Na+transport occur in the extracellular loops in OcHKT1;5-K, implying that crucial amino acid variation(s) in HKT1;5 have the potential to affect Na+transport characteristics. In the present study, we examined the diversities ofopen reading frame (ORF) in ninespecies (including homoeologous sequences from tetraploid) (Table S1) andexpression under salinity in wild and cultivatedspecies, and identified key complementary amino acid residue changes inHKT1;5-K/L homoeologous sequences that potentially control affinity for sodium. We also reported conserved orthologous gene sets between annotatedbacterial artificial chromosome (BAC) DNA harbouringand an analogous region on chromosome 1 ofssp.(IRGSP 1.0; https://rgp.dna.affrc.go.jp/IRGSP/Build2/ build2.html). Finally, weexamined HKT1;5 haplotype diversity in wild progenitor rice species and compared it with cultivated Asian rice accessions. Our data suggested that potentially new haplotypes present in the wild rice gene pool can be used to enhance the cultivatedgene pool for enhancing salinity tolerance.
RESULTS
OryzaHKT1;5 ORF features
HKT1;5 lengths in the nine examinedspecies range from 521 to 555 amino acid residues and show significant identity with previously reportedHKT1;5 sequences at NCBI. Thecoding region is spread across three exonic segments and the ends of Exons 1 and 2 are indicated by red arrows in Fig. S1. The predicted structure of HKT1;5 contains four transmembrane-P loop-transmembrane(MPM) motifs analogous to Ktr/Trk in bacteria/fungi (Su et al, 2015). A homology based 3D model for OsHKT1;5 has been proposed, with a similar MPM mode of organization (Cotsaftis et al, 2012). The S-G-G-G motif that constitutes the sodium SF in OsHKT1;5 (SF1, VSSM; SF2, NCGF; SF3, HSGE; SF4, NVGF) are absolutely conserved in allHKT1;5 examined (Cotsaftis et al, 2012; Waters et al, 2013; Su et al, 2015). Exon 1 ofencodes three pore helices (PH1‒PH3) and three selectivity filters (SF1‒ SF3), while Exon 2 encodes PH4and Exon 3 encodes SF4. In the fourth MPM domain, two conserved cysteine residues close to SF4have been predicted to control dimerization (Su et al, 2015) while adjacent arginine and lysine residues have been implicated in controlling free cation permeation (Kato et al, 2007). The four amino acid residues following SF4are absolutely conserved in allHKT1;5 sequences.
HKT1;5 sequences are organized into four main clades, in whichHKT1;5 sequences fromAA genome cluster together in subclade 1 (Clade 1), whileHKT1;5 sequences belonging to BB, BBCC and CC genome clusters in subclade 2 (Clade 1).(CCDD),(FF) and(KKLL) HKT1;5 sequences form three distinct clades (Fig. S2-A). WithinAA genome,andHKT1;5 sequences show the highest degree of relatedness (0.00480) whileHKT1;5 shows the highest genetic distance from otherHKT1;5 sequences examined (Fig. S2-B).
Ka/Ks (ratio of non-synonymous and synonymous substitutions) was computed for threeexonic regions. Ka/Ks is less than one for the 1st exonic region for all pairwise combinations acrossspecies, indicative of purifying selection (Fig. S2-C). Negative Tajima’sand Fu and Li’sstatistical scores for Exon 1 (Table S2) indicate low frequency polymorphisms in this region, reflective of the presence of highly conserved amino acid residues. For the 2nd and 3rd exons, 10 pairwise combinations ofAA genomesequences show non- synonymous substitutions (Ks = 0; Fig. S2-D and -E). For these AAgenome species, non-synonymous substitutions are indicative of positive selection.
Signatures of SNP conservation in HKT1;5 ORF are evident across Oryza species
Lineage specific SNPs and insertions are evident inORF sequences. Thus, allAA genomes show the presence of a characteristic 9-bp insertion (resulting in the addition of consecutive three amino acid residues ‘YSS’ as shown in Fig. S1). Also, onlyAA genome shows the presence of a ‘CTGATC’ sequence that codes for ‘LI’ (amino acid positions 320‒321 for,and; 321‒322 forand). Othersequences examined show the presence of ‘CGGCGG’ at the same position, which codes for ‘RR’. Both the ‘YSS’ insertion and the ‘LI’ change are seen inAA genomesequences occur in the region immediately following the MPM domains 1 and 2 (Exon 1), respectively.
ORFs from African cultivated rice.and its progenitor species., show distinct SNP conservation patterns from otherAA species (and). These include: four non-synonymous variations [S365 (AGC), R144 (CGC), P338 (CCG) and S508 (TCC)], three synonymous substitutions [T187 (ACT), A238 (GCA) and P298 (CCA)] and an insertion (GAC; coding for T204) (Fig. S3). This insertion of a codon results in one extra amino acid residue in theandHKT1;5 sequencesssp./andHKT1;5 sequences. The SNPs appear to be lineage specific based on our data and are also observed inandsequences reported at NCBI (nr and wgs databases; Table S3). Non-synonymous variations mostly occur in the intervening regions between the MPM domains or within structurally undefined regions within a given MPM domain (R144, R184 and S508). However, a few non-synonymous variations occur in structurally defined regions within a given MPM domain (T204, P338 and S365).
Negrão et al (2013) reported both synonymous and non-synonymous variations insequences. We also examined non-AA genomeORF sequences for presence or absence of non- synonymous SNPs inspecies (Table 1).BB genomesequences [(BB)/(BBCC)] share three non-synonymous substitutions G121 (GGT), R160 (CGC) and A305 (GCG), and the amino acid residues of G121 and R160 lie between helices D1m2and D2m1a, while A305 occurs in the D3m1helical region.
HKT1;5 expression under salinity treatment
With the exception ofand, allAA genome species showed upregulation ofexpression in leaf tissues under salinity. Inleaf tissue,expression levels were downregulated under salinity, and inleaves,expression levels were upregulated under salinity (Fig. 1).expression was significantly upregulated only in root tissues of,,and. In,expression in both leaf and root tissues appeared to be unaltered under salinity.

Table 1. Non-synonymous amino acid substitutions in O. sativa HKT1;5 sequences reported by Negrãoet al (2013) and comparison with those from Oryza species.
1,ssp.(AA);2,ssp.(AA);3,(AA);4,(AA);5,(AA);6,(BB);7,(BBCC);8,(CC);9,(CCDD);10,(FF);11,(KKLL).
Blue, green or yellow colours indicate conserved amino acid residues in relatedspecies. Corresponding amino acid residues insequences (landraces/varieties) reported by Platten et al (2013) are indicated in the last column. Amino acid changes highlighted in purple were identified in the 3K-rice genomes dataset. * indicates amino acid differences in OsHKT1;5 sequences reported by Ren et al (2005). ‘-’ indicates absence of the residue in specifiedspecies.

Fig. 1.expression inspecies in leaf and root tissues under control and salinity treated conditions (incremental salinity application).
Data are Mean ± SE for two biological replicates (each biological replicate was analyzed by qRT-PCR twice, each time in triplicate). Gene expression was quantified using the comparative CT (2-ΔΔCT) quantitation method with values representing ‘’-fold difference relative to the housekeeping control gene. Significance was calculated using the Student’s-test. **,< 0.01, ***,< 0.001 and ****,< 0.0001.
O. coarctata BAC clone Oc_Ba_0043B05 sequencing and annotation, and comparison of OcHKT1;5-K and -L orthologous regions
Shotgun sequencing ofBAC clone Oc_Ba_0043B05 produced 56 223 high quality reads, with a mean read length of 603 nucleotides and maximum modal read length of 772 bp (Table S4). About 98% of reads were assembled into 4 contigs, with the largest contig being 151 038 bp. Contig sequence was vector trimmed and the remaining 143 522bp sequences were repeat masked (37.2%; 53 390 bp). RepeatMasker annotation predicted 46 retroelements(31.2%; 1 L1/Cin4, 18 Ty1/and 27/DIRS1) and 37 DNA transposons (5.1%) (Table S5). Of the 26 predicted gene models (12 gene models on the coding strand and 14 on the non-coding strand) analyzed using BLASTN, 18 (77.0%) genes that had no any significant hits to cDNAs, expressed sequence tags or known proteins and were not conserved in other reportedgenomes were eliminated. Among the eight remaining gene models, five are known to be present inon chromosome 1. Exon/intron structures of the five genes were manually refined (Fig. 2). Of these, two gene models were supported bytranscriptome data (unpublished), while exon/intron structure of the third () was verified by qRT-PCR and cDNA cloning. The remaining three gene models are retrotransposons with similar organization reported from maize,and spinach.toin the annotatedBAC DNA clone (11 377‒117 064 bp) was compared with an analogous region (coordinates 11 350 487‒11 463 437 on chromosome 1) from().() contains an additional insertion of 7 263 bp, which includes two extra genes (betweenand;betweenand), suggesting insertion/deletion events have occurred in this region of chromosomal DNA duringspeciation.
A single syntenic block between the() locus (9.02 Mb‒15.38 Mb) and 143 kb BAC Oc_Ba_0043B05 sequence can be identified (Fig. S4). The exons 1 and 3 of-L and-K are roughly similar in length while the exon 2 differs in size (Fig. 3-A). OcHKT1;5-L (535 amino acid residues) is 14 amino acids longer than OcHKT1;5-K (521 amino acid residues), of which a nine amino acid residue insertion occurs between the 3rd and 4th SFs (Fig. 3-B). OcHKT1;5-L and -K homoeologous sequences show 81.2% identity and are more similar at their C-terminal ends.
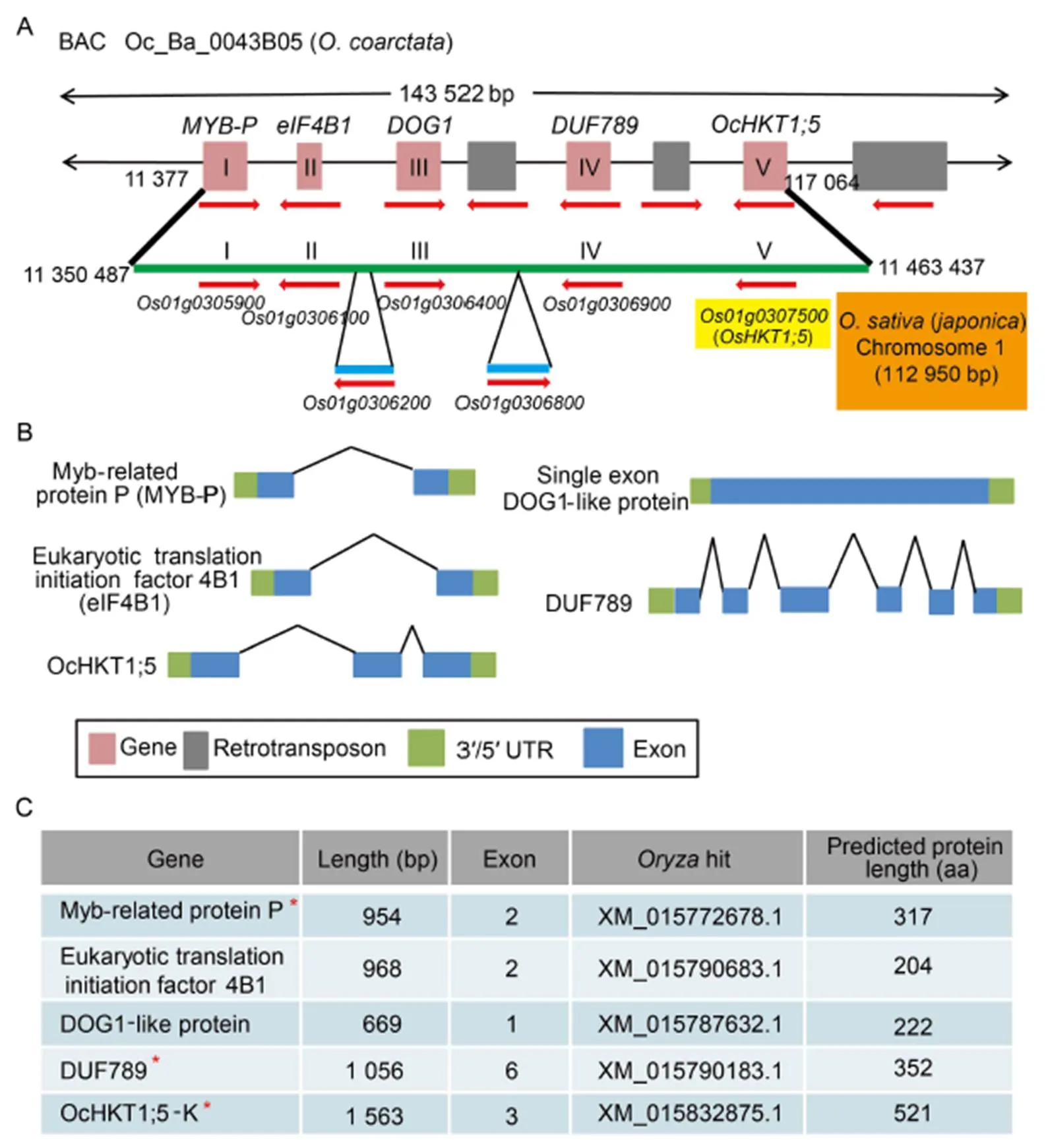
Fig. 2.BAC clone Oc_Ba_0043B05 sequencing and annotation.
A,Genomic alignment ofOc_Ba_0043B05 sequence with orthologousregion of() on chromosome 1.Predicted FGNESH gene models are indicated by pink boxes while retrotransposon gene models are indicated by grey boxes. Red arrows indicate gene orientation. Orthologous region inis indicated by a green line. Gene models I‒V are present. In addition, two gene insertions are also present in thesequence that are absent in the assembledBAC sequence.
B, Predicted exon/intron organizations of genemodels (I‒V) forOc_Ba_0043B05 sequence are conserved inrice.
C, Predicted protein lengths of models I‒V and accession numbers of correspondingtranscript orthologs. Asterisks indicate gene models validated intranscriptome or by qRT-PCR (unpublished).
Comparative homology modeling of OcHKT1;5-L
The OcHKT1;5-L model has four domain repeat structures,similar to the previously reported OcHKT1;5-K and OsHKT1;5 models (Cotsaftis et al, 2012; Somasundaram et al, 2020). Of the four key amino acid changes in OcHKT1;5-K occurring loops on the extracellular side, a positively charged residue near the ion pore entrance, K239, accounts for its lower affinity and higher Na+conductanceOsHKT1;5-Ni (Somasundaram et al, 2020). In comparison,OsHKT1;5-Ni has a negatively charged amino acid residue at the same position, conferring 16-fold higher affinity transport. Hence, amino acid residue level differences between OcHKT1;5-K and -L type models within a 2-nm radius of the pore region as well as transporter surface electrostatics were compared to understand their potential effects on ion conductance/affinity (Fig. 4). Key amino acid residues at the entrance such as E81, E83, G215, K216 and R217 are conserved between OcHKT1;5-K and -L. However, other residues including K239 and D314 in OcHKT1;5-K were found to be replaced by E242 and R338 in OcHKT1;5-L (Fig. 4-A and -B). This ‘swapping’ of oppositely charged residues would potentially ‘expand/increase’ the ion pore entrance, thereby enhancing its affinity for Na+transport. In addition, surface electrostatics suggested that the region around the pore entrance in OcHKT1;5-L was enriched in positively charged amino acid residues, while a similar region in OcHKT1;5-L was not so enriched in positively charged residues (including R338). This difference in surface electrostatics may have a regulatory role in controlling ion ‘approach’ (Fig. 4-C and -D).
HKT1;5 diversity in 3K-RG dataset
Seventy-six bi-allelic SNPs were identified in thesequence across 3K-RG dataset reported by Wang L et al (2018). Of these, eight are non- synonymous substitutions (Fig. 5). Seven non- synonymous substitutions occur in Exon 1 and one in Exon 2. Introns 1 and 2 show 46 and 12 SNPs, respectively. Non-synonymous SNPs result in the following amino acid changes insequence: (i) rs11463271C/T (Ser2Asn), (ii) rs11462894C/T (Asp128Asn), (iii) rs11462858G/C (Pro140Ala), (iv) rs11462792C/T (Val162Ile), (v) rs11462725C/T (Arg184His), (vi) rs11462282G/C (His332Asp), (vii) rs11462093G/C (Leu395Val), and (viii) rs11460344G/T (Gln429Lys) (Fig. S5). The eight non-synonymous SNPs identified are highlighted in Table 1.
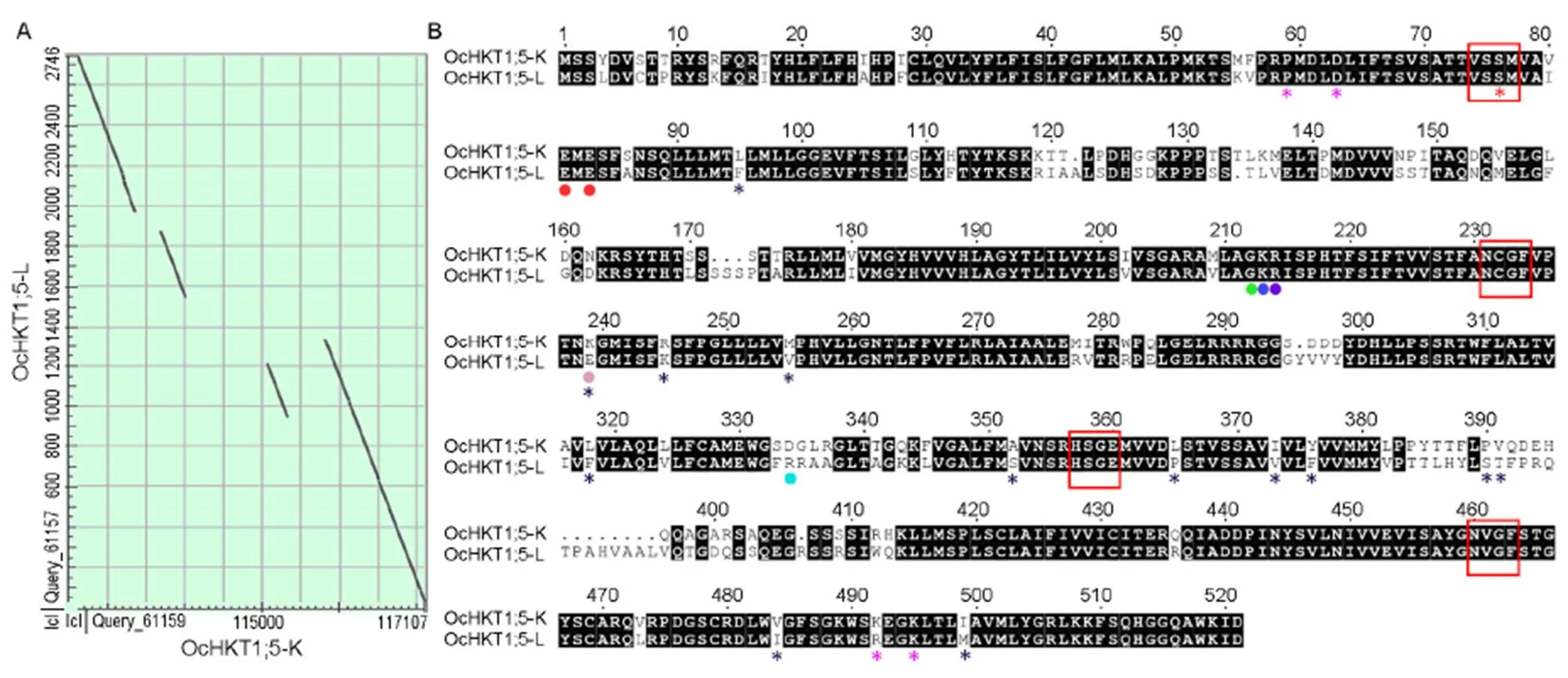
Fig. 3. Alignment ofHKT1;5 homoeologous genomic and protein sequences.
A, Dot plots showing four aligned regions ofOc_Ba_0043B05 sequence (143 522 bp) and-L genome (2 746 bp) sequences. BAC clone coordinates corresponding to four regions of homology (encompassing-K) with-L are indicated: (i) 115 810 to 117 107 (88% identity), (ii) 112 615 to 113 365 (87% identity), (iii) 115 067 to 115 333 (84% identity), and (iv) 113 686 to 114 009 (81% identity).
B, Alignment of OcHKT1;5-K (521 amino acid residues) and OcHKT1;5-L (535 amino acid residues) translated open reading fragments. The selectivity filter residues ‘S-G-G-G’ are marked by red asterisks while the selectivity filters are boxed in red. Conserved amino acid residues near the ion pore entrance crucial to ion transport are marked by a red dot. Amino acid residues indicated by red, green, dark blue or purple dots occur near the ion pore entrance and are the same in OcHKT1;5-K and OcHKT1;5-L. Resides marked by pink and light blue dots near the ion pore entrance show opposite charges in OcHKT1;5-K and OcHKT1;5-L. Pink asterisks indicate residues that are part of an extracellular putative cation coordination site. Black asterisks indicate residues that differ between OcHKT1;5-K and OcHKT1;5-L and either occur in transmembrane segments or in loops oriented towards the ion pore entrance.
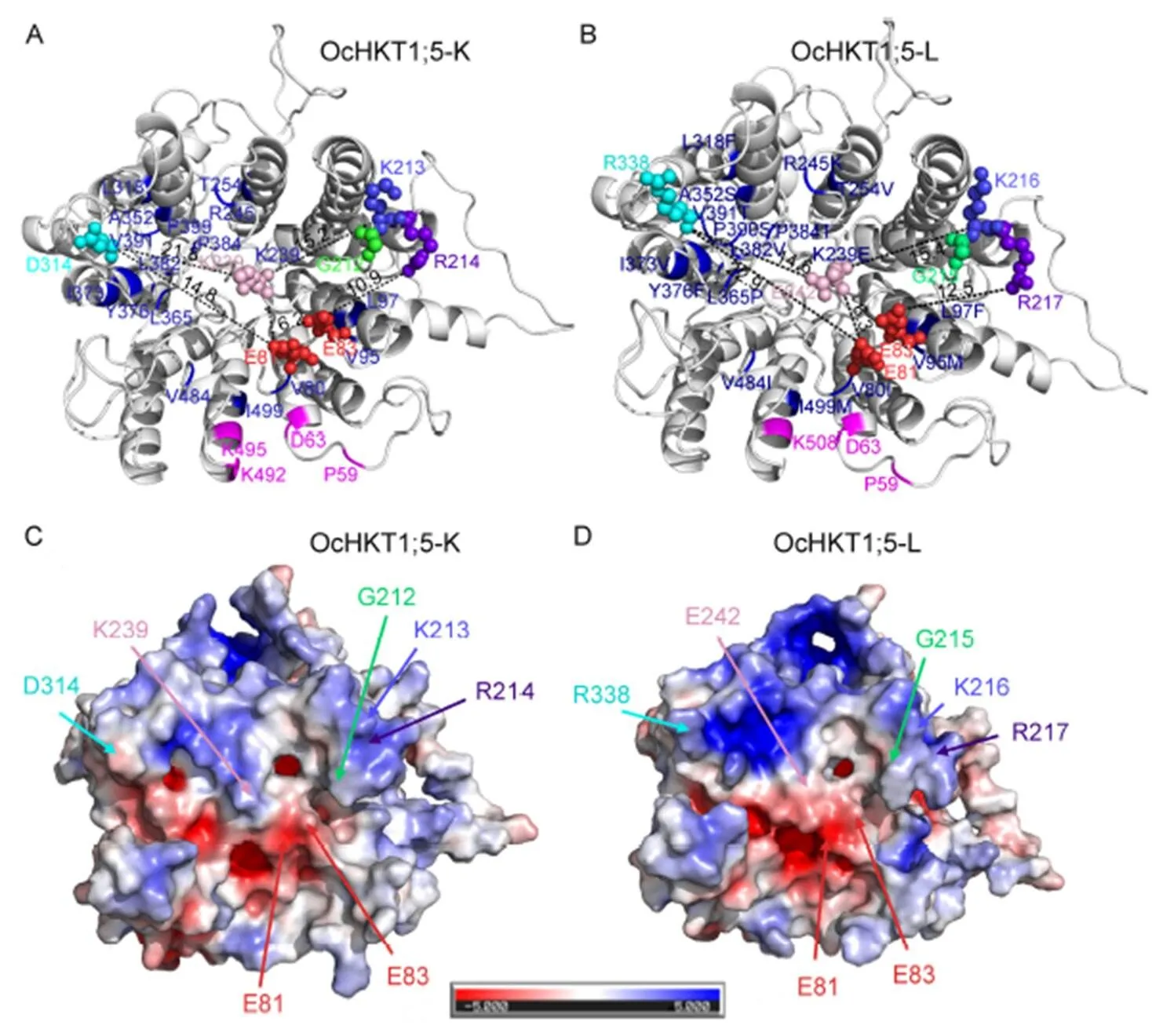
Fig. 4. Homology models and electrostatic maps of OcHKT1;5-K and OcHKT1;5-L proteins.
A and B, Model monomers of OcHKT1;5-K (A) and OcHKT1;5-L (B). Key residues at the pore entrance are highlighted as spheres, and the length between residues has been measured. Extracellular cation coordination sites (magenta) and residue differences within 2 nm from the pore region of both model (blue) are highlighted.
C and D, Comparative electrostatic maps of model generated for OcHKT1;5-K (C) and OcHKT1;5-L (D). Blue color indicates towards positive charge, while red color denotes negative charge. Majority of the residues at the pore region are negatively charged residues. Key amino acid residues or changes at the pore entrance have been marked.
3K-RG accessions have been classified into nine main and three admixed sub-populations (Wang W S et al, 2018). We mapped each non-synonymous SNP to the 12 sub- populations (Fig. S5). SNPs rs11463271 and rs11462894 are both C→T transversions, with the major ‘C’ allele (Ser2 and Asp128) distributed across all the 12 sub-populations, and the minor ‘T’ allele (Asn2 and Asn128) predominates in Ind (XI) and Aus (cA) (rs11463271) and Ind (XI) (rs11462894) categories, respectively. For SNP rs11462858, ‘G’ (Pro140) and ‘C’ (Ala140) alleles occur at almost equal frequencies in the 3K-RG population, with the ‘G’ allele occurring in the GJ, XI-3 and XI-adm sub-populations, and the ‘C’ allele occurring in the complementary XI, cA and cB sub- populations. The ‘C’ (Val162) allele is predominant across all the sub-populations for SNP rs11462792, and the extremely rare ‘T’ (Ile162) allele occurs only in the XI-3 and XI-adm categories (1.7% of 3K-RG population). For SNP rs11462725, the major ‘C’ (Arg184) allele occurs in all the sub-populations while the minor ‘T’ (His184) allele (20% of 3K-RG population) occurs near exclusively in the XI, cB and cA sub-populations. The minor ‘G’ allele for SNPs rs11462282 (Asp332) and rs11462093 (Val395) occurs primarily in the GJ accessions, while the major ‘C’ allele for SNPs rs11462282 (His332) and rs11462093 (Leu395) occurs in the XI, cA and cB sub-populations. For SNP rs11460344, the major ‘G’ (Gln429) allele is present in all the sub-populations while the minor ‘T’ (Lys429) allele occurs only in some XI and cA accessions.

Fig. 5. SNPs identified inin 3K-rice genomes (3K-RG).
A, Distribution of SNPs in thegenomic sequence in 3K-RG. SYN SNP, Synonymous substitutions in exonic regions (E1, E2 and E3); Non-SYN SNP, Non-synonymous substitutions in exonic regions; SNP (NC), SNPs in intronic regions (I1 and I2).
B, Circus based representation of SNPs and respective positions ingenomic sequence. SNP numbering as per IRGSP 1.0 (Chromosome 1). Outermost track represents SNPs and their respective positions (blue box at the top of the figure signifies the first polymorphic position; non-synonymous SNPs are indicated by orange boxes). Starting from outside, the second, third, fourth and fifth tracks represent the nucleotides adenine (red), thymine (blue), guanine (orange) and cytosine (green), respectively, with the height of each bar representing its frequency in the 3K-RG.
Cultivated rice HKT1;5 sequences (Wang L et al, 2018) were sorted into haplotypes for the eight predicted amino acid changes (Fig. 6-A and -B). Based on the 8 predicted amino acid changes in the OsHKT1;5 sequence, 10 haplotypes were identified that constitute 95% of the sequences analyzed (Fig. 6-B). The remainingsequences are heterozygous at one or more SNP positions in 3K-RG. Haplotypes 2 and 10 correspond to HKT1;5 sequences seen in the salt sensitive genotype Koshihikari (also Nipponbare, IRGSP 1.0) and salt tolerant genotype Nona Bokra (Ren et al, 2005), respectively, and differ from one another at four amino acid residues (P140A, R184H, H332D and V395L) (Fig. 6-B and -C). Haplotypes 2 and 8 show maximum number of accessions. Haplotypes 3 and 6 have the least number of accessions (three each). Haplotype 6 differs from Haplotype 10 at one amino acid residue (P140A), while Haplotypes 5 (P140A and R184H) and 8 (D128N and R184H) differ at two amino acid residues. Haplotypes 1 (P140A, V162I and R184H) and 3 (P140A, R184H and Q429K) differ at three amino acid residues from Haplotype 10. Haplotypes 4 (S2N, P140A, R184H and Q429K) and 9 (D128N, R184H, H332D and V395L) differ at four amino acid residues from Haplotype 10. Haplotype 7 differs from Haplotype 2 only at one amino acid residue. If Haplotypes 2 and 10 are considered as templates for ‘sensitive’ and ‘tolerant’ HKT1;5 sequences, then only Haplotype 7 groups with Haplotype 2 while Haplotypes 1, 3, 4, 5, 6, 8 and 9 group with Haplotype 10. Haplotypes 1 and 6 share the R184H amino acid residue change while the S2N amino acid residue change occurs only in Haplotype 4 and the Q429K amino acid residue change is seen in Haplotypes 3 and 4. P140A is seen in Haplotypes 8, 9 and 10. Consistent with the available population structure information, ‘salt sensitive HKT1;5’ Haplotype 2 (and related Haplotype 7) occur predominantly in the GJ () accessions while the ‘salt tolerant HKT1;5’ Haplotype 10 occurs in the XI () accessions as well as in the cA and cB groups. Haplotypes 1, 4, 5, 8 and 9 show a population distribution similar to Haplotype 10. Haplotype 1 is exclusively associated with the XI-3 sub-population. XI accessions occur majorly in Haplotype 8. Combining wild rice and cultivated rice HKT1;5 sequences for haplotyping analysis showed that wild rice HKT1;5 haplotypes occur in multiple nodes with cultivated rice accessions (Fig. 6-A). However, more importantly, there also occur nodes that are specific only for wild rice. This suggested that only a fraction of the wider wild rice HKT1;5 diversity has been passed onto cultivated rice.
Fig. 6. Haplotyping of HKT1;5 in wild and cultivated rice.
A, Only a subset of haplotypes seen in progenitor wild rice species (and) is seen in 3K-rice genomes (3K-RG).
B, Haplotypes recognized with reference to 8 non-synonymous amino acid residue changes in5 sequence and their distribution in 12 sub-populations (Wang W S et al, 2018) within 3K-RG [Amino acid changes indicted in bold are also referenced in Ren et al (2005)]. Haplotypes 2 (blue) and 10 (red) correspond to HKT1;5 sequences seen in salt sensitive genotype Koshihikari (also Nipponbare, IRGSP 1.0) and salt tolerant genotype Nona Bokra (Ren et al, 2005), respectively, and differ from one another in four amino acid residues indicated above. Occurrence of rare amino acid substitution in each haplotype is indicated in yellow. Haplotypes 11‒ 15 are heterozygous at one or more amino acid residue positions.
C, Haplotype network inferred for OsHKT1;5 for the 3K-RG dataset. Each circle represents a haplotype. and the size of each circle is proportional to haplotype frequency in the population.
DISCUSSION
Wild relatives of cultivated rice constitute an important reservoir for crop improve- ment as they are adapted to different biogeographic ranges as well as to a wide variety of biotic and abiotic conditions (Stein et al, 2018). While the sequence diversity of the sodium transporter HKT1;5 has been examined in cultivated rice(Negrão et al, 2013; Platten et al, 2013), its diversity inspecies has been examined to a limited extent (Mishra et al, 2016). The present study filled this gap. The present data reported additionalsequences from one diploid () and three tetraploidspecies (,and). Among these,is halophytic and saline tolerant (Bal and Dutt, 1986; Sengupta and Majumder, 2009; Rajakani et al, 2019).
Expansion of HKT1;5 ORF during Oryza evolution
TheORF appears to have undergone an expansion during the course ofgenome evolution, with two regions of theORF being sites of sequence insertions. The first major region in exon 1, comprising of multiple non-contiguous sequence blocks, occurs between regions coding for the first two MPM domains (between D1m2and D2m1a) (Fig. S1) and occurs exclusively inAA genomesequences. This region is predicted to be intracellular and shows a serine rich tract that may be subject to post-translational modifications (Negrão et al, 2013). Similarly, the second minor region with sequence insertion occurs in exon 2 and precedes the region coding for the fourth MPM domain. This second region occurs inBB, CC and BBCC genomes and is absent fromAA genomesequences. On the other hand, stretches coding for HKT1;5MPM domains are relatively conserved. Exon 1 incodes for three of four MPM domains and sequence stretches that occur between the MPM domain coding regions are sites of insertions or deletions. These include the nine nucleotide insertions (coding for ‘YSS’) in exon 1 ofsequences, a 6-bp change (CTGATC) in exon 2 ofAA genomes. In addition, lineage specific changes in African cultivated rice,and its progenitor species,, also occur in the intervening regions between the MPM domains or within structurally undefined regions within a given MPM domain.
HKT1;5 expression in Oryza species under salinity
Root-specific upregulation ofis associated with salinity tolerance in(Ren et al, 2005; Prusty et al, 2018). Our data suggested thatexpression in roots is upregulated in(FL478; salinity tolerant),(),,,andwhile down- regulated in(Nona Bokra),under salinity. Prusty et al (2018) reported root-specific upregulation ofexpression in both salinity tolerant and sensitivecultivars as well asCCDD species under salinity. Our data suggested that root-specificexpression in the sensitive(IR28) is unchanged under salinity and reduced in(CCDD). The salinity regime utilized in this study was planned to account for the ionic effect of Na+onexpression.is expressed in phloem of diffuse vascular bundles in basal nodes of Dongjin rice and prevent Na+transfer to young leaf blades, thereby protecting leaf blades fromsalt stress injury in the growing phase (Kobayashi et al, 2017). The expression data here, upregulation ofexpression in leaf blades of mostAA species that may have an additional role in thespecies and is worth exploring further.
Genomic regions encompassing OcHKT1;5-K show additional genes in more evolved Oryza genomes
homoeologous sequences from allotetraploid halophytic wild rice(KKLL) are reported here. Bothtranslated ORFs (Homeologs K and L; Fig. 3) show closer identity to(81%‒87%)..is a more ancestral species (Mondal et al, 2018; Stein et al, 2018) that diverged from.around 10 millions years ago (MYA). Closer identity of both homoeologoussequences withis consistent with this. TheHKT1;5 evolutionary tree placessequence intermediate betweenand other members of thecomplex (,and).B-C genome split occurred about 4.8 MYA, with CCDD tetraploids arising 0.9 MYA and BBCC tetraploids arising 0.3‒0.6 MYA (Wang et al, 2009). Annotation of the 143 kb BAC clone (with theHKT1;5-K homoeolog) and comparison with correspondingssp.(IRGSP 1.0) sequence suggested that two additional genes are present in, resulting in an additional 7 263 bp sequence insertion. Analysis (https://plants.ensembl.org/) suggested that the two insertion/deletion events have occurred independently in time and in different lineages duringgenome evolution. Thus,orthologs occur in allAA genomes examined and also in(BB genome) on chromosome 1, but not in(FF). Nevertheless,orthologs occur only inAA genome on chromosome 1, suggesting that this gene insertion/deletion event is relatively recent and probably originates prior to diversification ofAA genome from other ancestralspecies. In othergenomes [(AA),(BB) and(FF)], the closest homolog is present on chromosome 8 (ONIVA08G11000, OPUNC08G09620 and OB08G- 18970; https://plants.ensembl.org/).
Comparative homology models for OcHKT1;5-K and -L identify complementary amino acid residues in OcHKT1;5-L that potentially enhance affinity for Na+ transport
Modeling of homoeologous OcHKT1;5 structures suggested that OcHKT1;5-L may have a higher affinity for Na+transport, compared to the previously characterized OcHKT1;5-K that attributes to ‘swapping’of positive and negatively charged amino acid residues at the ion pore entrance. In addition, a cluster of positively charged residues in the vicinity of the pore entrance in OcHKT1;5-L may constitute one of the many regions that ions pass through before entering the pore. A positively charged surface in the vicinity of the ion pore may have a ‘repelling’ effect on Na+ions, directing them towards the ion pore. This differs from the negatively extracellular cation coordination site identified in OsHKT2;2 that appears to modify ion transport kinetics by sequestering ions briefly before movement towards the ion pore (Riedelsberger et al, 2019). Further mutagenesis of amino acid residues identified in OcHKT1;5-L coupled with transport studies will have to be carried out to confirm their importance in controlling affinity for Na+and/or kinetics of transport.
japonica rice shows lesser HKT1;5 diversity compared to indica rice
Ten HKT1;5 haplotypic groups have been identified in the 3K-RG dataset on the basis of non-synonymous substitutions. Among these, Haplotypes 2 and 10 correspond to HKT1;5 sequences in the salt sensitive genotype Koshihikari and salt tolerant genotype Nona Bokra, respectively. Both these haplotypes constitute a significant fraction of the 3K-RG dataset and appear to occur in mutually exclusive GJ () and XI () accessions, respectively. In addition, Haplotype 10 also has a significant number of(cA) and aromatic (cB) accessions. Further, Haplotype 8 that constitutes another significant number in the 3K-RG dataset differs from Haplotype 10 at two amino acid residues. Haplotype 8 also occurs inaccessions (Fig. 6-B). Analysis of data for 529 accessions reported by Yang et al (2018) shows a similar distribution of(Haplotype 2 in this study) andaccessions (Haplotypes 10 and 8 in this study). Finally, Haplotype 10 differs from Haplotypes 6, 5, 8, 1 and 3 at 1‒3 amino acid residues and all haplotypes occur in predominantlyaccessions. However, only Haplotype 7 differs from Haplotype 2 and both occur predominantly only inaccessions. Reduced haplotypic diversity of HKT1;5 inrice pools in this analysis as well as that by Yang et al (2018) are indicative of reduced diversity within the founding wild rice pools that contribute to their origin. At any rate, increased HKT1;5 haplotype diversity in//aromatic accessions would have been derived independently during multiple domestication events involving geographically distinct wild rice pools originally containing the diverse haplotypes, similar to theandgenes inaccessions (Civáň and Brown, 2017; Wang W S et al, 2018).
The geographical provenances of Haplotypes 10 and 2 are partially overlapping (Fig. S6). Multiple independent domestications of Asian ricehave contributed to,andsub- populations (Huang et al, 2012; Civáň et al, 2015; Wang W S et al, 2018). Evidence suggests that aromatic rice is a result of hybridization betweenand, with tropical and temperate versions ofconstituting later adaptations of one crop (Civáň et al, 2019). Presence of Haplotype 2 in both temperate and tropicalsub-populations suggests that the haplotype occurred prior to diversification ofsub-population. Sensitive and tolerant HKT1;5 Haplotypes 2 and 10 have evolved independently inas well asandaccessions, respectively. Haplotype 10 may have been transferred into the aromatic sub-population from ansub-population, based on the proposed origin of the aromatic rice sub-population (Civáň et al, 2019). The predominance of specific HKT1;5 haplotypes in the 3K-RG dataset suggested a possible ecophysiological advantage conferred in a given climatic, edaphic and geographical context by a given HKT1;5 haplotypic combination analogous toAtHKT1and AtHKT1alleles (Busoms et al, 2018).
Mishra et al (2016) explored only wild rice within a defined geographical area of the Indian subcontinent. A more comprehensive exploration of HKT1;5 diversity in more geographically diverse areas may identify additional HKT1;5 haplotypes that can be future determinants of salt tolerance.
METHODS
Plant materials and genomic DNA extraction
Eight wildspecies (one accession per species) along with African cultivated rice () were selected, representative of seven genome types (Table S1). Propagules of halophytic ricewere obtained from Pichavaram, Tamil Nadu, India. Genomic DNA was isolated from leaves ofspecies using the CTAB method (Doyle and Doyle, 1987), quantified using Multiskan™ GO Microplate Spectrophotometer v.3.2 (Thermo Scientific, USA) and also checked by agarose gel electrophoresis.
PCR amplification and sequencing
Primers (Table S6, primer pairs 1‒6) corresponding to the three exonic regions ofwere designed to amplify DNA fragments from genomic DNA regions ofspecies listed in Table S1 using proof reading DNA polymerase. PCR reactions were carried out in a reaction volume of 20 μL with appropriate genomic DNA (50 ng), 0.5 μmol/L each of specific forward and reverse primers, 2.5 mmol/L dNTPs, proof reading PrimeSTAR GXL DNA Polymerase (2.5 U) (TaKaRa, USA). PCR cycling conditions were as follows: 30 cycles of DNA denaturation (98 ºC, 10 s), annealing (60 ºC, 15 s), extension (68 ºC, 1‒2 min). Amplified products were analyzed by agarose gel electrophoresis, purified by gel extraction (NucleoSpin Gel and PCR Cleanup, Macherey-Nagel, USA). Eluted PCR products were sequenced (Eurofins, India) using both forward and reverse gene specific primers (Table S6). Subsequently, specific primers were designed to obtain completegenomic information for thespecies listed above as overlapping fragments (Table S7).
Sequence analysis
Chromatogram reads (for PCR products) were verified manually and analyzed by BLAST. The ORF within each assembledsequence was identified based on alignment withsequences reported in the NCBI database.ORFs were translated using EXPASY (Gasteiger et al, 2003) and aligned using ConSurf (Ashkenazy et al, 2016). MEGA-X (Kumar et al, 2018) was used to assess pairwise distances and determine phylogenetic relatedness among the 11 HKT1;5 sequences while DnaSP v 5.0 (Librado and Rozas, 2009) was used to estimate nucleotide diversity.
O. coarctata BAC clone Oc_Ba_0043B05: Sequencing and annotation
sequence fragments were isolated by PCR fromgenomic DNA (primers used are listed in Table S6). Based on PCR fragment sequences, 92BAC DNA clones were obtained from Arizona Genomics Institute, USA [an extendedgenomic region (± 250 kb) centered aroundwas used for comparison], representing orthologous regions of interest for each of theallotetraploid sub-genomes (KKLL). PCR analysis was used to identify one clone for each of thesub-genomes (KK and LL) that contained the completeorthologous region (L sub-genome Oc_Ba_0027O13/ A9and K sub-genome Oc_Ba_0043B05/B1). Oc_Ba_0027O13/ A9 BAC DNA was isolated and using specifically designed PCR primers (generating overlapping fragments), theL homoeolog was PCR amplified as overlapping fragments from Oc_Ba_0027O13/A9 BAC DNA using a proof reading polymerase and sequenced (Table S6). The sequence of the largest contig was analyzed by MegaBLAST to trim vector sequence (pAGIBAC) (Ammiraju et al, 2006).
The assembledBAC DNA sequence (Oc_Ba_0043B05) was repeat masked with an inbuilt Dfam curatedTE library (RepeatMasker 4.0.6). The repeat masked sequence was annotated usingprediction program FGENESH 2.6.ssp.Group was used as a reference (Solovyev et al, 2006). Annotated and predicted candidate genes were assigned based on presence of a defined functional domain and availability of a homolog outside of. The annotated Oc_Ba_0043B05 sequence (143 522 bp) is deposited at NCBI.
O. coarctata BAC clone Oc_Ba_0043B05: Synteny analysis
MegaBLAST and BLASTN were used to align-L and -K homoeologous sequences.-L (2 746 bp) was queried against-K (143 522 bp) using default parameters by BLASTN. Synteny analysis was performed using SyMAP (Soderlund et al, 2011). IRGSP 1.0 was used as a reference with default parameters.
Homology modeling of OcHKT1;5-L
OcHKT1;5-K structure has been modeled previously using crystal structures reported for bacterial potassium transporters KtrAB from(PDB ID: 4J7C) and TrkH from(PDB ID: 3PJZ) (Somasundaram et al, 2020). The same templates were used to model homoeologous OcHKT1;5-L. Secondary structures of KtrAB, TrkH and OcHKT1;5-L sequences were aligned using PsiPred (Jones, 1999). OcHKT1;5-L showed 20.3% identity with TrkH and was used to model the N- and C-terminal regions, while KtrAB showing 19.6% sequence identity was used to construct the core regions of the query protein using Modeller v9.12 (Šali and Blundell, 1993). Twenty models were generated with five loop refinement runs per model. The model with the lowest Discrete Optimized Protein Energy score was selected for energy minimization using the Optimized Potentials for Liquid Simulations force field. The modeled structure was validated using MolProbity for Ramachandran allowed conformations (Williams et al, 2018) and ProSA (Wiederstein and Sippl, 2007). Quality checks using the Ramachandran Map showed 93.7% of the residues were in the allowed regions and 79.7% in favorable regions (Fig. S7-A). Quality of the model was also confirmed using ProSA (Fig. S7-B). The output model was structurally superimposed on the previously generated OcHKT1;5-K model (Somasundaram et al, 2020). Electrostatic potential differences were calculated using Adaptive Poisson-Boltzmann Solver (Baker et al, 2000) and visualized using Pymol (DeLano, 2002). The previously modeled OcHKT1;5-K and the currently reported OcHKT1;5-L model showed a good degree of agreement, based on a root mean square deviation value of 0.233.
RNA isolation and qRT-PCR expression
Seeds ofssp.cvs.Pokkali, Nona Bokra, FL478 (salt tolerant), IR28 (salt sensitive),ssp.,,,,,,,,,andwere incubated at 50 ºC for 5 d and incubated at 28 ºC in dark conditions for germination (5‒10 d). Germinated seeds were transferred to Yoshida medium for one month and subjected to incremental salinity treatment (25, 50, 75 and 100 mmol/L NaCl) at a 3-d interval. Leaf and root tissues were frozen for RNA isolation. Total RNA was isolated using RNAiso Plus (TaKaRa, USA). cDNA was synthesized from 1 μg of DNaseItreatedleaf and root total RNA.
First strand cDNA synthesis was performed in a 20 µL reaction volume with 1 µg of total RNA and Superscript III (Invitrogen, USA) at 42 ºC for 60 min, followed by heat inactivation at 70 ºC for 10 min. Primer pair HKT8_W9 Fwd/ HKT8_W9 Rev (5′-ATTCTGGCTCCAACTGCTGTACT-3′/ 5′-GTGAAGATCAGGTCCAAGTCCAT-3′) was used for allspecies exceptssp.. For the latter, primer pair HKT8_JP Fwd/HKT8_JP Rev (5′-TCTGGATGCC ACTACTCCTAGATATG-3′/5′-AAACCCAAGAGGGAGATGAAGAG-3′) was used, andwas used as an internal control. qRT-PCR (Quant Studio 6; Thermo Fisher, USA) was carried out in a reaction volume of 10 µL [1 µL of cDNA, 5 µL of 2× TB GreenTMPrimix ExTMII (TaKaRa, USA), 0.5 µL each of a given primer pair with final concentration of 250 nmol/L] under the following cycling conditions: 95 ºC (30 s), 40 cycles of denaturation at 95 ºC (5 s), annealing and extension at 60 ºC (30 s) in a 96-well optical reaction plate (Thermo Fisher, USA). Each qRT-PCR reaction was performed in triplicate to evaluate data reproducibility for two biological replicates. Amplicon specificity was verified by melt curve analysis (60 ºC–95 ºC at 40 cycles). Gene expression was quantified using the comparative CT (2-ΔΔCT) quantitation method with values representing ‘n’-fold difference relative to housekeeping control.
OsHKT1;5 diversity in progenitor wild rice, 3K-RG and haplotyping
gene sequences from progenitor wild rice species (Mishra et al, 2016) and(Platten et al, 2013) were mapped onto a Nipponbare chr01 reference (IRGSP-1.0) using Geneious R6 (https://geneious.com). The resulting alignment containing thegene was extracted and highly diverged sequences were removed, keeping only,andsequences together with(MN150526.1) and(MN164536.1)sequences as an outgroup for further analysis. This alignment was complemented with diversity data from the 3K-RG project (https://snp-seek.irri.org/_snp.zul) as follows. First, the SNP- seek database was queried with the IRGSP-1.0 genomic coordinates of thegene (chr01: 11 458 954‒11 463 442), using the ‘3Kall’ SNP set, which yielded 322 polymorphic sites. Then, using the genomic coordinates of the reference, the alignment of the gene sequences and the SNP data were merged, retaining all polymorphic positions found in the coding sequence of the gene. Positions polymorphic in the gene alignment but absent from the 3K-RG SNP matrix were retained and assumed to carry the reference variant in all 3K-RG accessions. Subsequently, positions with singletons were removed, and all accessions with ambiguous calls (indicating heterozygotes) were deleted, giving a dataset with 40 polymorphic sites within 2 877 accessions. These data were used to construct a Median-Joining network using Network 5 (Bandelt et al, 1999). For analysis of non-synonymous SNPs with, bi-allelic non-synonymous SNPs were downloaded from the SNP-seek database. Heterozygotic genotypes were excluded from 3K-RG dataset and remaining genotypes sorted into haplotypic groups (using Java version 1.7 based script) and the haplotyping network inferred manually.
ACKNOWLEDGEMENTS
This study was supported by the Department of Biotechnology, Government of India (Grant No. BT/PR11396/NDB/52/118/ 2008) and Council for Scientific and Industrial Research, India for Senior Research Fellowship (Grant No. 09/656(0018)/ 2016-EMR-1) to Shalini Pulipati.The authors thank Dr. Jose Luis Goicoechea at Arizona Genomics Institute, USA for providing assistance in selecting theBAC clones, Directors in Central Rice Research Institute, Cuttack, India and International Rice Research Institute, the Philippines for providingseeds, and National Center for Biological Sciences, India for infrastructural facilities. R. Sowdhamini acknowledges funding and support provided by JC Bose Fellowship (Grant No. SB/S2/JC-071/2015) from Science and Engineering Research Board, India and Bioinformatics Centre Grant funded by Department of Biotechnology, India (Grant No. BT/PR40187/BTIS/137/9/2021).
SUPPLEMENTAL DATA
The following materials are available in the online version of this article at http://www.sciencedirect.com/journal/rice-science; http://www.ricescience.org.
Fig. S1. Alignment ofHKT1;5 sequences.
Fig. S2. Phylogenetic analysis of;5 sequences, cladogram of HKT1;5 sequences fromspecies based on protein sequences prepared using MEGA X pairwise divergence and percent identity betweensequences, and Ka/Ks ratios for threeexonic segments for orthologous pairs fromspecies.
Fig. S3. Multiple alignment ofandsequences (specific regions) generated in this study withsequence fromAA genomes retrieved from NCBI.
Fig. S4. Synteny mapping of() locus andBAC Oc_Ba_0043B05 sequence.
Fig. S5. Distribution of non-synonymous SNPs in 3K-RG sub- populations.
Fig. S6. Country-wise geographical provenances for Haplotypes 2 and 10 for HKT1;5 in 3K-RG (based on non-synonymous substitutions) are partially overlapping.
Fig. S7. OcHKT1;5-L structure quality validation.
Table S1.wild rice species and correspondingsequences reported in this study.
Table S2. Summary statistics ofsequence variation and Tajima’sinspecies.
Table S3.sequences (1‒11) or,,,andwgs (12‒19) retrieved from NCBI.
Table S4.BAC clone Oc_Ba_0043B05 assembly statistics.
Table S5. Transposable elements present in assembled and vector trimmedBAC clone Oc_Ba_0043B05.
Table S6. Primer pairs used for amplificationexonic regions fromspecies.
Table S7. Primer pairs used to obtain complete genomic information forfromspecies.
Ali A, Raddatz N, Aman R, Kim S, Park H C, Jan M, Baek D, Khan I U, Oh D H, Lee S Y, Bressan R A, Lee K W, Maggio A, Pardo J M, Bohnert H J, Yun D J. 2016. A single amino-acid substitution in the sodium transporter HKT1 associated with plant salt tolerance., 171(3): 2112‒2126.
Almeida P M F, de Boer G J, de Boer A H. 2014. Assessment of natural variation in the first pore domain of the tomato HKT1;2 transporter and characterization of mutated versions ofexpressed inoocytes and via complementation of the salt sensitivemutant., 5: 600.
Ammiraju J S S, Luo M Z, Goicoechea J L, Wang W M, Kudrna D, Mueller C, Talag J, Kim H, Sisneros N B, Blackmon B, Fang E, Tomkins J B, Brar D, MacKill D, McCouch S, Kurata N, Lambert G, Galbraith D W, Arumuganathan K, Rao K, Walling J G, Gill N, Yu Y, SanMiguel P, Soderlund C, Jackson S, Wing R A. 2006. Thebacterial artificial chromosome library resource: Construction and analysis of 12 deep-coverage large-insert BAC libraries that represent the 10 genome types of the genus., 16(1): 140‒147.
An D, Chen J G, Gao Y Q, Li X, Chao Z F, Chen Z R, Li Q Q, Han M L, Wang Y L, Wang Y F, Chao D Y. 2017.drives adaptation ofto salinity by reducing floral sodium content.,13(10): e1007086.
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. 2016. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules., 44: W344‒W350.
Asins M J, Villalta I, Aly M M, Olías R, Alvarez D E Morales P, Huertas R, Li J, Jaime-Pérez N, Haro R, Raga V, Carbonell E A, Belver A. 2013. Two closely linked tomato HKT coding genes are positional candidates for the major tomato QTL involved in Na+/K+homeostasis., 36(6): 1171‒1191.
Baker N, Holst M, Wang F. 2000. Adaptive multilevel finite element solution of the Poisson-Boltzmann equation: II. Refinement at solvent-accessible surfaces in biomolecular systems., 21(15): 1343‒1352.
Bal A R, Dutt S K. 1986. Mechanism of salt tolerance in wild rice (Roxb)., 92: 399‒404.
Bandelt H J, Forster P, Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies., 16: 37–48.
Busoms S, Paajanen P, Marburger S, Bray S, Huang X Y, Poschenrieder C, Yant L, Salt D E. 2018. Fluctuating selection on migrant adaptive sodium transporter alleles in coastal., 115(52): E12443‒E12452.
Byrt C S, Xu B, Krishnan M, Lightfoot D J, Athman A, Jacobs A K, Watson-Haigh N S, Plett D, Munns R, Tester M, Gilliham M. 2014. The Na+transporter, TaHKT1;5-D, limits shoot Na+accumulation in bread wheat., 80: 516‒526.
Civáň P, Brown T A. 2017. Origin of rice (L.) domestication genes., 64: 1125‒1132.
Civáň P, Craig H, Cox C J, Brown T A. 2015. Three geographically separate domestications of Asian rice., 1: 15164.
Civáň P, Ali S, Batista-Navarro R, Drosou K, Ihejieto C, Chakraborty D, Ray A, Gladieux P, Brown T A. 2019. Origin of the aromatic group of cultivated rice (L.) traced to the Indian subcontinent., 11(3): 832‒843.
Cotsaftis O, Plett D, Shirley N, Tester M, Hrmova M. 2012. A two-staged model of Na+exclusion in rice explained by 3D modeling of HKT transporters and alternative splicing., 7: e39865.
DeLano W L. 2002. Pymol: An open-source molecular graphics tool. http://www.ccp4.ac.uk/newsletters/newsletter40/11_pymol.pdf.
Doyle J J, Doyle J L. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue., 19: 11‒15.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel R D, Bairoch A. 2003. ExPASy: The proteomics server for in-depth protein knowledge and analysis., 31: 3784‒3788.
Hoang T, Tran T, Nguyen T, Williams B, Wurm P, Bellairs S, Mundree S. 2016. Improvement of salinity stress tolerance in rice: Challenges and opportunities., 6(4): 54.
Houston K, Qiu J E, Wege S, Hrmova M, Oakey H, Qu Y, Smith P, Situmorang A, Macaulay M, Flis P, Bayer M, Roy S, Halpin C, Russell J, Schreiber M, Byrt C, Gilliham M, Salt D E, Waugh R. 2020. Barley sodium content is regulated by natural variants of the Na+transporter HvHKT1;5., 3(1): 258.
Huang X H, Kurata N, Wei X H, Wang Z X, Wang A H, Zhao Q, Zhao Y, Liu K Y, Lu H Y, Li W J, Guo Y L, Lu Y Q, Zhou C C, Fan D L, Weng Q J, Zhu C R, Huang T, Zhang L, Wang Y C, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan X P, Xu Q, Dong G J, Zhan Q L, Li C Y, Fujiyama A, Toyoda A, Lu T T, Feng Q, Qian Q, Li J Y, Han B. 2012. A map of rice genome variation reveals the origin of cultivated rice., 490: 497‒501.
Jiang Z L, Song G S, Shan X H, Wei Z Y, Liu Y Z, Jiang C, Jiang Y, Jin F X, Li Y D. 2018. Association analysis and identification of ZmHKT1;5 variation with salt-stress tolerance, 9: 1485.
Jones D T. 1999. Protein secondary structure prediction based on position-specific scoring matrices., 292(2): 195‒202.
Kato N, Akai M, Zulkifli L, Matsuda N, Kato Y, Goshima S, Hazama A, Yamagami M, Guy H R, Uozumi N. 2007. Role of positively charged amino acids in the M2Dtransmembrane helix of Ktr/Trk/HKT type cation transporters., 1(3): 161‒171.
Kobayashi N I, Yamaji N, Yamamoto H, Okubo K, Ueno H, Costa A, Tanoi K, Matsumura H, Fujii-Kashino M, Horiuchi T, Nayef M A, Shabala S, An G, Ma J F, Horie T. 2017.mediates Na+exclusion in the vasculature to protect leaf blades and reproductive tissues from salt toxicity in rice., 91(4): 657‒670.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms., 35: 1547‒1549.
Kumar V, Singh A, Mithra S V A, Krishnamurthy S L, Parida S K, Jain S, Tiwari K K, Kumar P, Rao A R, Sharma S K, Khurana J P, Singh N K, Mohapatra T. 2015. Genome-wide association mapping of salinity tolerance in rice ()., 22(2): 133‒145.
Librado P, Rozas J. 2009. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data., 25: 1451‒1452.
Menguer P K, Sperotto R A, Ricachenevsky F K. 2017. A walk on the wild side:species as source for rice abiotic stress tolerance., 40: 238‒252.
Mishra S, Singh B, Panda K, Singh B P, Singh N, Misra P, Rai V, Singh N K. 2016. Association of SNP haplotypes offamily genes with salt tolerance in Indian wild rice germplasm., 9: 15.
Mondal T K, Rawal H C, Chowrasia S, Varshney D, Panda A K, Mazumdar A, Kaur H, Gaikwad K, Sharma T R, Singh N K. 2018. Draft genome sequence of first monocot-halophytic speciesreveals stress- specific genes., 8: 13698.
Moradi F, Ismail A M, Gregorio G B, Egdane J A. 2003. Salinity tolerance of rice during reproductive development and associationwith tolerance at the seedling stage., 8: 105‒116.
Munns R, James R A, Xu B, Athman A, Conn S J, Jordans C, Byrt C S, Hare R A, Tyerman S D, Tester M, Plett D, Gilliham M. 2012. Wheat grain yield on saline soils is improved by an ancestral Na+transporter gene.,30(4): 360‒364.
Negrão S, Almadanim M C, Pires I S, Abreu I A, Maroco J, Courtois B, Gregorio G B, McNally K L, Oliveira M M. 2013. New allelic variants found in key rice salt-tolerance genes: An association study., 11(1): 87‒100.
Platten J D, Cotsaftis O, Berthomieu P, Bohnert H, Davenport R J, Fairbairn D J, Horie T, Leigh R A, Lin H X, Luan S, Mäser P, Pantoja O, Rodríguez-Navarro A, Schachtman D P, Schroeder J I, Sentenac H, Uozumi N, Véry A A, Zhu J K, Dennis E S, Tester M. 2006. Nomenclature for HKT transporters, key determinants of plant salinity tolerance., 11(8): 372‒374.
Platten J D, Egdane J A, Ismail A M. 2013. Salinity tolerance, Na+exclusion and allele mining ofinand: Many sources, many genes, one mechanism?, 13: 32.
Prusty M R, Kim S R, Vinarao R, Entila F, Egdane J, Diaz M G Q, Jena K K. 2018. Newly identified wild rice accessions conferring high salt tolerance might use a tissue tolerance mechanism in leaf., 9: 417.
Rajakani R, Sellamuthu G, Saravanakumar V, Kannappan S, Shabala L, Meinke H, Chen Z H, Zhou M X, Parida A, Shabala S, Venkataraman G. 2019. Microhair on the adaxial leaf surface of salt secreting halophyticRoxb. show distinct morphotypes: Isolation for molecular and functional analysis., 285: 248‒257.
Ren Z H, Gao J P, Li L G, Cai X L, Huang W, Chao D Y, Zhu M Z, Wang Z Y, Luan S, Lin H X. 2005. A rice quantitative trait locus for salt tolerance encodes a sodium transporter., 37: 1141‒1146.
Riedelsberger J, Vergara-Jaque A, Piñeros M, Dreyer I, González W. 2019. An extracellular cation coordination site influences ion conduction of OsHKT2;2., 19: 316.
Rus A, Yokoi S, Sharkhuu A, Reddy M, Lee B H, Matsumoto T K, Koiwa H, Zhu J K, Bressan R A, Hasegawa P M. 2001. AtHKT1 is a salt tolerance determinant that controls Na+entry into plant roots., 98: 14150‒14155.
Rus A, Lee B H, Muñoz-Mayor A, Sharkhuu A, Miura K, Zhu J K, Bressan R A, Hasegawa P M. 2004. AtHKT1 facilitates Na+homeostasis and K+nutrition in planta., 136(1): 2500‒2511.
Šali A, Blundell T L. 1993. Comparative protein modelling by satisfaction of spatial restraints., 234(3): 779‒815.
Sengupta S, Majumder A L. 2009. Insight into the salt tolerance factors of a wild halophytic rice,: A physiological and proteomic approach., 229(4): 911‒929.
Soderlund C, Bomhoff M, Nelson W M. 2011. SyMAP v3.4: A turnkey synteny system with application to plant genomes., 39(10): e68.
Solovyev V, Kosarev P, Seledsov I, Vorobyev D. 2006. Automatic annotation of eukaryotic genes, pseudogenes and promoters., 7(Suppl 1): S10.1-12.
Somasundaram S, Véry A A, Vinekar R S, Ishikawa T, Kumari K, Pulipati S, Kumaresan K, Corratgé-Faillie C, Sowdhamini R, Parida A, Shabala L, Shabala S, Venkataraman G. 2020. Homology modeling identifies crucial amino-acid residues that confer higher Na+transport capacity of OcHKT1;5 fromRoxb., 61(7): 1321‒1334.
Stein J C, Yu Y, Copetti D, Zwickl D J, Zhang L, Zhang C J, Chougule K, Gao D Y, Iwata A, Goicoechea J L, Wei S, Wang J, Liao Y, Wang M H, Jacquemin J L, Becker C, Kudrna D, Zhang J W, Londono C E M, Song X, Lee S, Sanchez P, Zuccolo A, Ammiraju J S S, Talag J, Danowitz A, Rivera L F, Gschwend A R, Noutsos C, Wu C C, Kao S M, Zeng J W, Wei F J, Zhao Q, Feng Q, Baidouri M E, Carpentier M C, Lasserre E, Cooke R, da Rosa Farias D, da Maia L C, dos Santos R S, Nyberg K G, McNally K L, Mauleon R, Alexandrov N, Schmutz J, Flowers D, Fan C Z, Weigel D, Jena K K, Wicker T, Chen M S, Han B, Henry R, Hsing Y C, Kurata N, de Oliveira A C, Panaud O, Jackson S A, Machado C A, Sanderson M J, Long M Y, Ware D, Wing R A. 2018. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus., 50(2): 285‒296.
Su Y, Luo W G, Lin W H, Ma L Y, Kabir M H. 2015. Model of cation transportation mediated by high-affinity potassium transporters (HKTs) in higher plants., 17(1): 1‒3.
The 3 000 rice genomes project. 2014. The 3 000 rice genomes project., 3: 7.
Thomson M J, de Ocampo M, Egdane J, Rahman M A, Sajise A G, Adorada D L, Tumimbang-Raiz E, Blumwald E, Seraj Z I, Singh R K, Gregorio G B, Ismail A M. 2010. Characterizing thequantitative trait locus for salinity tolerance in rice., 3(2/3): 148‒160.
van Bezouw R F H M, Janssen E M, Ashrafuzzaman M, Ghahramanzadeh R, Kilian B, Graner A, Visser R G F, van der Linden C G. 2019. Shoot sodium exclusion in salt stressed barley (L.) is determined by allele specific increased expression of., 241: 153029.
Véry A A, Nieves-Cordones M, Daly M, Khan I, Fizames C, Sentenac H. 2014. Molecular biology of K+transport across the plant cell membrane: What do we learn from comparison between plant species?, 171(9): 748‒769.
Wang B S, Ding Z Y, Liu W, Pan J, Li C B, Ge S, Zhang D M. 2009. Polyploid evolution incomplex of the genus., 9(1): 250.
Wang L, Liu Y H, Feng S J, Wang Z Y, Zhang J W, Zhang J L, Wang D, Gan Y T. 2018.gene regulating K+state in whole plant improves salt tolerance in transgenic tobacco plants., 8: 16585.
Wang W S, Mauleon R, Hu Z Q, Chebotarov D, Tai S S, Wu Z C, Li M, Zheng T Q, Fuentes R R, Zhang F, Mansueto L, Copetti D, Sanciangco M, Palis K C, Xu J L, Sun C, Fu B Y, Zhang H L, Gao Y M, Zhao X Q, Shen F, Cui X, Yu H, Li Z C, Chen M L, Detras J, Zhou Y L, Zhang X Y, Zhao Y, Kudrna D, Wang C C, Li R, Jia B, Lu J Y, He X C, Dong Z T, Xu J B, Li Y H, Wang M, Shi J X, Li J, Zhang D B, Lee S, Hu W S, Poliakov A, Dubchak I, Ulat V J, Borja F N, Mendoza J R, Ali J, Li J, Gao Q, Niu Y C, Yue Z, Naredo M E B, Talag J, Wang X Q, Li J J, Fang X D, Yin Y, Glaszmann J C, Zhang J W, Li J Y, Hamilton R S, Wing R A, Ruan J, Zhang G Y, Wei C C, Alexandrov N, McNally K L, Li Z K, Leung H. 2018. Genomic variation in 3 010 diverse accessions of Asian cultivated rice., 557: 43‒49.
Waters S, Gilliham M, Hrmova M. 2013. Plant high-affinity potassium (HKT) transporters involved in salinity tolerance: Structural insights to probe differences in ion selectivity., 14(4): 7660‒7680.
Wiederstein M, Sippl M J. 2007. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins., 35: W407‒W410.
Williams C J, Headd J J, Moriarty N W, Prisant M G, Videau L L, Deis L N, Verma V, Keedy D A, Hintze B J, Chen V B, Jain S, Lewis S M, Arendall W B 3rd, Snoeyink J, Adams P D, Lovell S C, Richardson J S, Richardson D C. 2018. MolProbity: More and better reference data for improved all-atom structure validation., 27(1): 293‒315.
Yang C W, Zhao L, Zhang H K, Yang Z Z, Wang H, Wen S S, Zhang C Y, Rustgi S, von Wettstein D, Liu B. 2014. Evolution of physiological responses to salt stress in hexaploid wheat., 111(32): 11882‒11887.
Yang M, Lu K, Zhao F J, Xie W B, Ramakrishna P, Wang G Y, Du Q Q, Liang L M, Sun C J, Zhao H, Zhang Z Y, Liu Z H, Tian J J, Huang X Y, Wang W S, Dong H X, Hu J T, Ming L C, Xing Y Z, Wang G W, Xiao J H, Salt D E, Lian X M. 2018. Genome-wide association studies reveal the genetic basis of ionomic variation in rice., 30(11): 2720‒2740.
23 January 2021;
17 May2021
Gayatri Venkataraman (gayatri@mssrf.res.in)
Copyright © 2022, China National Rice Research Institute. Hosting by Elsevier B V
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
Peer review under responsibility of China National Rice Research Institute
http://dx.doi.org/10.1016/j.rsci.2021.12.003
(Managing Editor: Wu Yawen)
杂志排行

Rice Science的其它文章
- Rice Drying, Storage and Processing: Effects of Post-Harvest Operations on Grain Quality
- UvWhi2 Is Required for Stress Response and Pathogenicity in Ustilaginoidea virens
- Fine Mapping of QTLs for Stigma Exsertion Rate from Oryza glaberrima by Chromosome Segment Substitution
- Simple Bioassay for PAMP-Triggered Immunity in Rice Seedlings Based on Lateral Root Growth Inhibition
- Ionomic Profiling of Rice Genotypes and Identification of Varieties with Elemental Covariation Effects
- Cold Plasma: A Potential Alternative for Rice Grain Postharvest Treatment Management in Malaysia