Prudent application of single-cell RNA sequencing in understanding cellular features and functional phenotypes in cancer studies
2022-01-17XuanhaoXuZeminZhang
Xuanhao Xu ,Zemin Zhang,2
1Beijing Advanced Innovation Center for Genomics,Peking-Tsinghua Center for Life Sciences,Peking University,Beijing 100871,China;2Biomedical Pioneering Innovation Center (BIOPIC) and School of Life Sciences,Peking University,Beijing 100871,China
Abstract This decade has seen remarkable advances in the field of high-throughput single cell techniques.Single-cell RNA sequencing (scRNA-seq) has proven to be a powerful strategy to study the heterogeneity in clinical samples,providing an unbiased approach to uncover the characteristics in different cell subsets.To ensure the reproducibility and robustness of biological discoveries,researchers need to be aware of hidden caveats in tissue dissociation,cell capturing and transcripts measurement which may affect cell composition assessment and cellular function annotation.With measured interpretation of data and innovations in experimental and technical approaches,scRNA-seq can greatly unravel the heterogeneity in complex system and improve our understandings in tissue homeostasis and cancer biology.
Keywords:Single-cell RNA sequencing;tumor microenvironment;immune cells;cancer study
Introduction
Solid tumors have complex cellular composition of stroma cells,fibroblasts,infiltrated immune cells and extracellular components.The tumor microenvironment (TME) is highly heterogeneous,which correlates with cancer evolution,metastasis and the efficacy in clinical treatment.For cancer studies and therapeutic applications,it is crucial to understand the cell composition,functional phenotype and tumor heterogeneity (1).Recent advances in various single-cell sequencing approaches have proven their tremendous abilities in the characterization of transcriptional profiles,immunophenotypes,lineage origins and epigenetic landscape in individual cells.The most efficient and widely applied method is single-cell RNA sequencing (scRNA-seq),which enables quantification of transcripts inside each captured cell.It has made major breakthroughs in the understanding of complex biological samples,especially in the comprehensive analysis of cell composition and transcriptional signatures,thus revealing novel cell subsets with distinct functional phenotypes in depth.
As scRNA-seq is a delicate combination of various biological techniques and complex computational pipelines,it has more critical steps that need to be carefully handled than conventional approaches.In this perspective,we will illustrate notable caveats for scRNA-seq data interpretation that are pertinent to the cell quantitative assessment and functional phenotype annotation.We will also use several scRNA-seq studies of cancer immunology as examples to summarize strategies to circumvent these caveats and extract new biological insights.
Prudence in quantitative assessment of cells by scRNA-seq
scRNA-seq offers a high-resolution view to assess the detailed cell composition in tumor tissues.However,none of the available dissociation methods uniformly capture all cell types in a given sample at the same efficiency.Nonimmune cells are often associated with cell-cell adhesion and cytoskeletal interaction systems which make them more resistant to physical treatment and enzymatical digestion than those free immune cells.In addition,due to the cell-cell junction and extracellular components,nonimmune cells are also easily aggregated and filtered out before being loaded on single cell capturing platforms.There may also be sensitive cell populations like granulocytes,especially neutrophils,which are short-lived but important in tumor progression and metastasis.They are rich in proteases inside and express only a few hundred genes,making them more prone to being lost during the cell suspension preparation and mRNA capture procedure.Furthermore,studies focusing on peripheral blood mononuclear cells (PBMCs) will expectedly deplete the neutrophil populations since PBMCs are in the middle layer with very few neutrophils after density gradient centrifugation (2).Moreover,clinical samples themselves are highly heterogeneous,causing batch effects for both biological and technical replicates.Due to the additional intratumoral heterogeneity,the resection area of tumor samples may contribute to significant variations of cellular proportions in the single cell suspension.As a result,rare cell types may not be consistently captured.
Aside from the sample preparation complications,cell shapes and sizes are often limiting factors for most single cell platforms,which might selectively remove certain cells while preferentially retaining other cells.Lymphocytes,for example,are typically easier to capture than fibroblast cells due to their smaller and regular sizes.Consequently,it is not advisable to directly compare the fractions of different cell types quantitatively,e.g.T cell levelsvs.neutrophil levels.Instead,it is much more reliable to compare the distribution preference of the same cell types across different clinical samples that are processed with the same experimental protocols.Under the identical procedures,multiple dissection areas (e.g.tumor core,tumor edge,adjacent normal tissue) can be compared to aid the study of intratumoral differences (3).For example,for colorectal cancer and hepatocellular carcinoma,although the absolute cell percentages varied across tumor samples,statistical analyses showed that exhausted T cells were specifically found in tumor tissues while central memory and effector T cells were preferentially present in the blood (3,4).
Since scRNA-seq has become more commercially and technically available in recent years,researchers are inclined to collect a larger sample cohort,which aids the robust recovery and analysis of various cell populations.Furthermore,the pre-enrichment of those sensitive and rare cell populations with magnetic beads or cell sorting can be used to augment the quantitative detection of such cells while minimizing the signal from unwanted cell mixture (4).In addition,during the preparation of cell suspension,to better retain thebona fidecell populations in solid samples,optimization of dissociation protocols as well as gentle procedures is preferred if physical treatment and enzymatic digestions affect the cell viability and functional states (Figure 1).
Interpretation of diverse functional phenotypes revealed by scRNA-seq
A central aspect of single cell analysis is the identification of cell types and the elucidation of functional states.Such analyses are heavily dependent on the processing of cellgene expression matrices,which contain quantitative counting of all captured transcripts for each cell in theory.However,in practice,due to the low RNA amount from an individual cell and random RNA expression,as well as variable gene capture efficiencies in sequencing,some genes may be undetectable in one cell but maintain a moderate or low expression level in another cell of the same type.This dropout phenomenon exists in all single cell platforms but is more notable in the unique molecular identifier (UMI)-based droplet-seq compared with the full length-based switching mechanism at 5’ end of the RNA transcript (SMART-seq) approach. Although most abundant genes are captured,the dropout effect does weaken the precision of cell type identification and functional annotation if critical signature genes happen to be absent.
To tackle the dropout and batch effects,experimental and computational pipelines have been developed in recent years to improve the interpretation of single cell data,such as the widely used toolkit Seurat (5).Meanwhile,each single cell protocol or platform holds its own advantages.Droplet-based methods capture a larger scale of cells but with smaller gene detection numbers,whereas the full length-based method can better interrogate molecular features inside each cell at the cost of lower throughput and more intensive labor.With an appropriate data integration procedure,this combined approach can achieve an extensive cellular landscape in-depth (Figure 1).Recent studies of colorectal cancer and hepatocellular carcinoma(HCC) applied both SMART-seq2 and 10× Genomics platforms,thereby recovering specific macrophages and under-characterized dendritic cells (DCs) as well as some intermediate subsets (3,4).
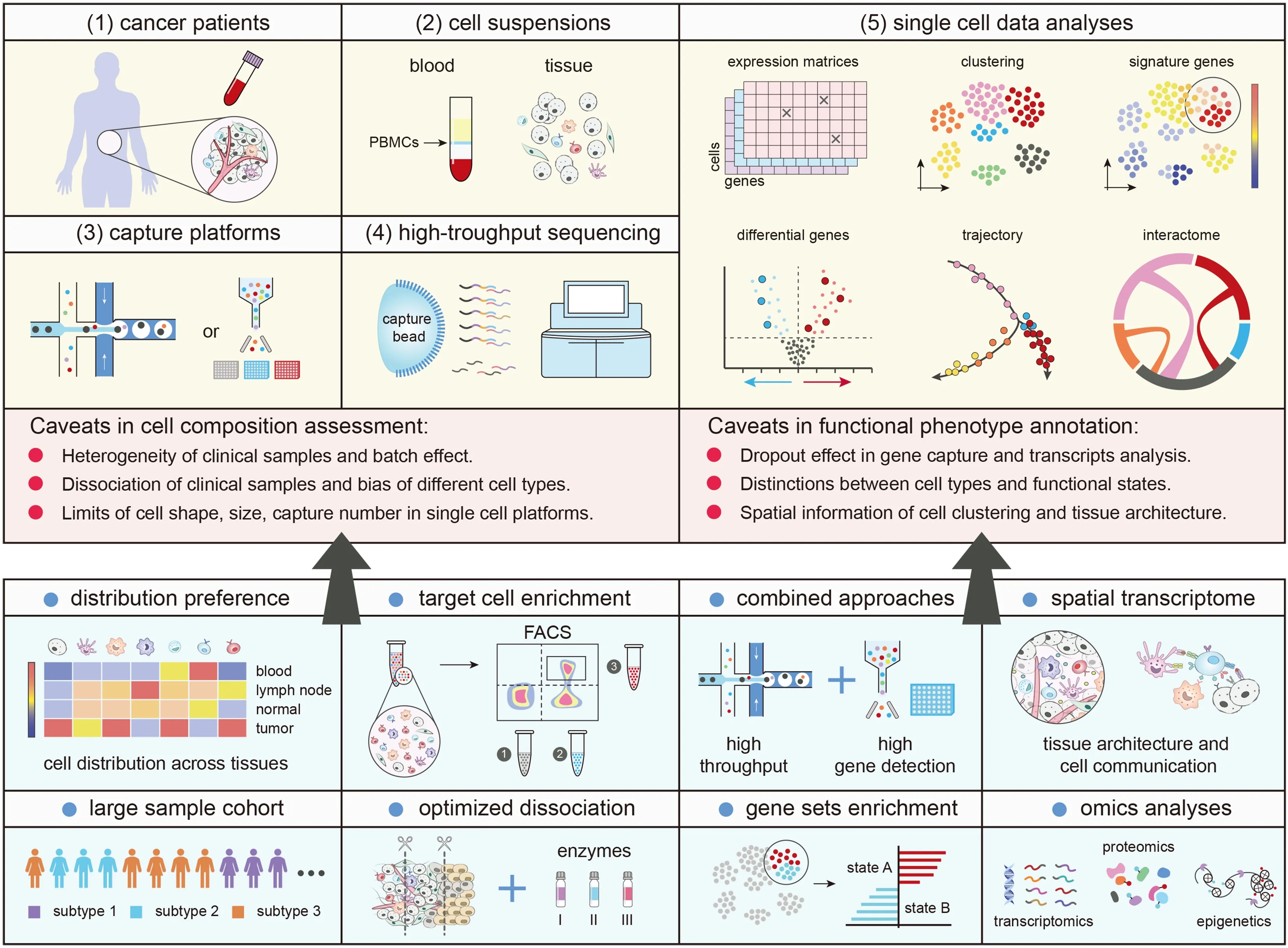
Figure 1 Illustration of scRNA-seq and strategies to better assess cell composition and elucidate cell phenotypes.Caveats in scRNA-seq implicate tissue dissociation,cell capturing (left panel) and transcripts measurement (right panel) which may affect cell composition assessment and cellular function annotation.Strategies for solutions can be divided into two parts.1) Section of cell assessment (left panel):with classified tumor subtypes and tissue origins,the cell distribution preference can be better evaluated.Meanwhile,pre-enrichment of target cells helps augment sensitive or rare cell signals from unwanted cell mixture and achieve relatively reliable composition results.Optimized dissociation protocols and gentle treatment alleviate damage to cells and retain populations during single cell suspension preparation;and 2) section of functional phenotype annotation (right panel):combination of the droplet-based and the full length-based platform increases the number of cell recovery and improves the precision of cell state annotation.Analyses of gene sets,pathway enrichment and transcriptional factors assist a better interrogation of cellular states and phenotypes.In addition,spatial transcriptomics are necessary to illustrate cell communications and tissue architectures.Furthermore,the integration of omics data helps scrutinize cellular phenotypes in-depth and establish cell atlas with multiple scales.scRNA-seq,single-cell RNA sequencing;PBMC,peripheral blood mononuclear cell;FACS,fluorescence activated cell sorting.
Differentially expressed genes provide the most obvious clue for interrogating distinct cellular phenotypes.Here we take T cell as an example.The infiltration and activation of T cells can be critical for the prognostic prediction in the cancer treatment,while the understanding of their heterogeneity,clonal expansion and dynamic changes can also point to new therapeutic strategies.In an HCC study,scRNA-seq revealed distinctive functional compositions of T cells in tumor or peripheral sites.Analysis of signature genes showed that exhausted T cells expressed high levels ofPDCD1,CTLA4andHAVCR2while it also specifically expressedLAYN,which was not previously reported (6).This feature is also consistent in the subsequent studies across various cancer types that implicateLAYN,encoding a cell surface protein,to be a potentially interfering target for T cell rejuvenation and the anti-tumor treatment.
Since cells are delicately orchestrated by the gene regulation network,it is important to further annotate cellular states and phenotypes through detailed analysis of gene sets,pathway enrichment and transcriptional factors.Cell cycle,for example,is considered as one of the confounding factors during cluster analyses.In an HCC study,gene sets associated with cell cycle (such asMCM7,STMN1andMKI67) were used to help discover those minor sub-clusters from the major T exhaustion populations (6).It underscored the necessity to investigate different phenotypes and to distinguish cell states from cell clusters.Another tumor-infiltrating cell group,myeloid cells,play key roles in the regulation of immune activities and the cancer progression.A study of colorectal cancer carefully examined tumor associated macrophage (TAM)populations and clustered them intoC1QC+TAM andSPP1+TAM subtypes,with distinctive roles suggested (4).Transcriptional factor analyses showed specific expressions ofMAF/MAFBandFOS/JUNinC1QC+TAMs,whileCEBPBandZEB2were notable inSPP1+TAMs.Further investigation into cellular phenotype revealed thatC1QC+TAMs were enriched in the pathways of complement activation,antigen processing and presentation.By contrast,SPP1+TAMs showed a strong enrichment of tumor angiogenesis and tumor vasculature signals,thus indicating a pro-tumorigenic function.Furthermore,a pancancer study provided additional proof for the existence of these cell populations across different tumors (7).Overall,the analysis of cell compositions and cellular phenotypes can be fairly robust based on the scRNA-seq data.
In addition,to further enable the illustration of molecular features and cellular phenotypes,trajectory analyses based on the transcriptional continuity and clonal relationship can be used to predict cell dynamics during biological processes.Furthermore,since most cells are organized in tissues and communicate via membrane proteins and secreted molecules to fine tune their phenotypes,ligand-receptor interactome profiles can provide additional clues in the regulation axis,cell recruitment and cellular functions.The aforementioned HCC study highlighted an under-characterized cluster of DCs,LAMP3+DC,using a combination of trajectory and interaction analyses.It appeared thatLAMP3+DC cells,arising from conventional DCs,might migrate from tumors to hepatic lymph nodes,and they further displayed interaction potentials with T cell populations (exhausted T cells,proliferative T cells and regulatory T cells) through interleukin-15 and the co-stimulatorCD28/B7family (3).
Trends of scRNA-seq in cancer studies
One of the drawbacks of the current scRNA-seq technologies is that critical information of tissue architecture is lost when clinical samples are dissociated.Recent developed technologies,such as spatial transcriptomics,present an attractive solution for studying cellular compositions and functional phenotypes while maintaining their spatial information.This also enables the comparative analysis of cells across different sample regions,and facilitates the study of cell-cell interactions in the TME (8).Together with transcriptional profiles,spatial positioning of certain cell clusters can be pinpointed on tumor samples,thus revealing the distribution preference of different cell types in the normal or malignant regions.
Additional technologies aimed at revealing the epigenetic or the chromosomal state of single cells could effectively complement the expression-based methods.For example,assay for transposase-accessible chromatin with single cell sequencing (scATAC-seq) enables the study of cellular states by identifying the accessible or closed genome regions.It can directly confirm the existence of unique regulatory programs across cell types beyond transcriptomes.Furthermore,scATAC-seq can explore mechanisms that drive phenotype differences via the analyses of transcription factor activity,cis-elements accessibility and enhancer-promoter connections.The active signals ofEOMESandTBX21,for example,are present in both natural killer and T cell populations,while lineage-determining factorTCF7is restricted in T cells (9).The distinct features of the transcriptomic and epigenetic profiles make these two broad approaches complimentary,since their combined usage compensates for the data perturbation in individual methods,leading to better elucidation of cell functional states.
In addition to individual and focused studies,mega projects like the Human Cell Atlas project and the Human Tumor Atlas Network initiatives are charging forward.Collectively,with the accumulation of various clinical samples and clear profiles of cell types and molecular features,the integrated single cell atlas will help identify novel cellular states,therapeutically relevant cell subsets,predictive biomarkers,as well as cell interactome across the tissue and tumor backgrounds,thereby elucidating TME,estimating immunotherapy responses and explaining drug resistance mechanisms during clinical application (1,10).
Conclusions
The growth of scRNA-seq studies is explosive in recent years and a plethora of cell populations have been classified and scrutinized across tumors. These studies have generated an impressive number of high-quality datasets,revealing complex cell compositions and transcriptional patterns in the tumor development.Despite unavoidable caveats associated with single cell sequencing,researchers are becoming more comfortable and experienced in avoiding those pitfalls while still extracting novel biological insights from the complex single cell data.Further integration of scRNA-seq together with spatial methods and multi-omics data will not only help discover those salient features of tissue architecture in healthy and pathological tissues,but also help advance the overall understanding of cancer biology and ultimately the therapeutic strategies.The high-resolution single cell data have already provided unprecedented depth in understanding the complex TME,and it will likely reach to a point in the future when clinical samples will be routinely analyzed by single cell technologies for diagnostic and therapeutic purposes.
Acknowledgements
This study is supported by the National Natural Science Foundation of China (No. 81988101,91942307,31991171);Beijing Municipal Science and Technology Commission (No. Z201100005320014) and the Postdoctoral Fellowship of Peking-Tsinghua Center for Life Sciences.
Footnote
Conflicts of Interest:Zhang Z is a founder and scientific advisor for Analytical Biosciences.The other author has no conflicts of interest to declare.
杂志排行

Chinese Journal of Cancer Research的其它文章
- Novel technologies in cfDNA analysis and potential utility in clinic
- ZNF292 suppresses proliferation of ESCC cells through ZNF292/SKP2/P27 signaling axis
- Development and validation of a questionnaire-based risk scoring system to identify individuals at high risk for gastric cancer inChinese populations
- Associations of centralization with health care quality for gastric cancer patients receiving gastrectomy in China
- Identification of lymph node metastasis by computed tomography in early gastric cancer
- Deep learning model improves radiologists’ performance in detection and classification of breast lesions