Novel technologies in cfDNA analysis and potential utility in clinic
2022-01-17JieLiMengyueXuJunyaPengJingqiaoWangYupeiZhaoWenmingWuXunLan
Jie Li ,Mengyue Xu ,Junya Peng ,Jingqiao Wang ,Yupei Zhao,4,5 ,Wenming Wu,5 ,Xun Lan
1Department of Basic Medical Sciences,School of Medicine,Tsinghua University,Beijing 100084,China;2Department of General Surgery,Peking Union Medical College Hospital,Chinese Academy of Medical Science &Peking Union Medical College,Beijing 100730,China;3Department of Medical Research,Peking Union Medical College Hospital,Chinese Academy of Medical Science &Peking Union Medical College,Beijing 100730,China;4 Tsinghua-Peking Joint Center for Life Sciences,Tsinghua University,Beijing 100084,China;5 Department of State Key Laboratory of Complex Severe and Rare Diseases,Peking Union Medical College Hospital,Chinese Academy of Medical Science and Peking Union Medical College,Beijing 100730,China
Abstract The profiling of plasma cell-free DNA (cfDNA) is becoming a valuable tool rapidly for tumor diagnosis,monitoring and prognosis.Diverse plasma cfDNA technologies have been in routine or emerging use,including analyses of mutations,copy number alterations,gene fusions and DNA methylation.Recently,new technologies in cfDNA analysis have been developed in laboratories,and potentially reflect the status of epigenetic modification,the immune microenvironment and the microbiome in tumor tissues.In this review,the authors discuss the principles,methods and effects of the current cfDNA assays and provide an overview of studies that may inform clinical applications in the near future.
Keywords:cfDNA;liquid biopsy;cancer diagnosis;recurrence monitoring;therapy response
Introduction
Molecular characterization of tumors has revolutionized the field of precise oncology,with genomic profiling strategies guiding treatment selection for multiple cancer types (1,2).Traditionally,molecular profiling uses tumor tissues derived from tumor biopsy or surgical resection,with the disadvantages of invasiveness (3),a lack of realtime monitoring (4,5) and regional limitations (6).More recently,liquid biopsies,particularly cell-free DNA(cfDNA) from plasma,have emerged as important supplementary tools to standard biopsy (7-10).
Plasma cfDNA refers to fragmented DNA presents in the noncellular component of the blood,which has been released through cell apoptosis or necrosis (11).Notably,cfDNA is usually 150-200 base pairs in length (4,8,12) and presents at a concentration of 10-15 ng per milliliter (8) in the plasma of healthy persons,with a half-life shorter than 2 h (4,8).Plasma cfDNA originates from the death of multiple cell types,such as hematopoietic cells and histiocytic cells (4).Specifically,cfDNA released from tumor cells is named circulating tumor DNA (ctDNA).
In tumor tissues,variances have been observed in the cellular composition and molecular status compared to healthy controls.Cellular composition refers to the subtypes and numbers of tumor cells,immune cells,stromal cells and microbiome (13,14).The molecular status differs in various factors,including genomic,transcriptomic and epigenetic variances (15-17).Effective characterization of these variances may potentially be translated into clinical practice in the early screening,diagnosis and prognosis of patients with tumors (18).Earlier plasma ctDNA analyses used next-generation sequencing (NGS) to assess somatic alterations (including mutations,copy number alterations,gene fusions and DNA methylation) (3,19-24) and were in routine clinical use with commercially available tests.Currently,new cfDNA tests,including specific fragment patterns (25),transcription start site (TSS) coverage (5),T cell receptor sequencing (26) and associated microbiome cfDNA analysis (27),have been developed in the laboratory(Figure 1).These cfDNA techniques effectively reflect genomic variance,epigenetic modification,microenvironment interaction and the associated microbiome status of tumor tissues,which show potency in clinical translation.In this review,we will discuss these emerging techniques of plasma cfDNA assays and their potential clinical applications in the near future.
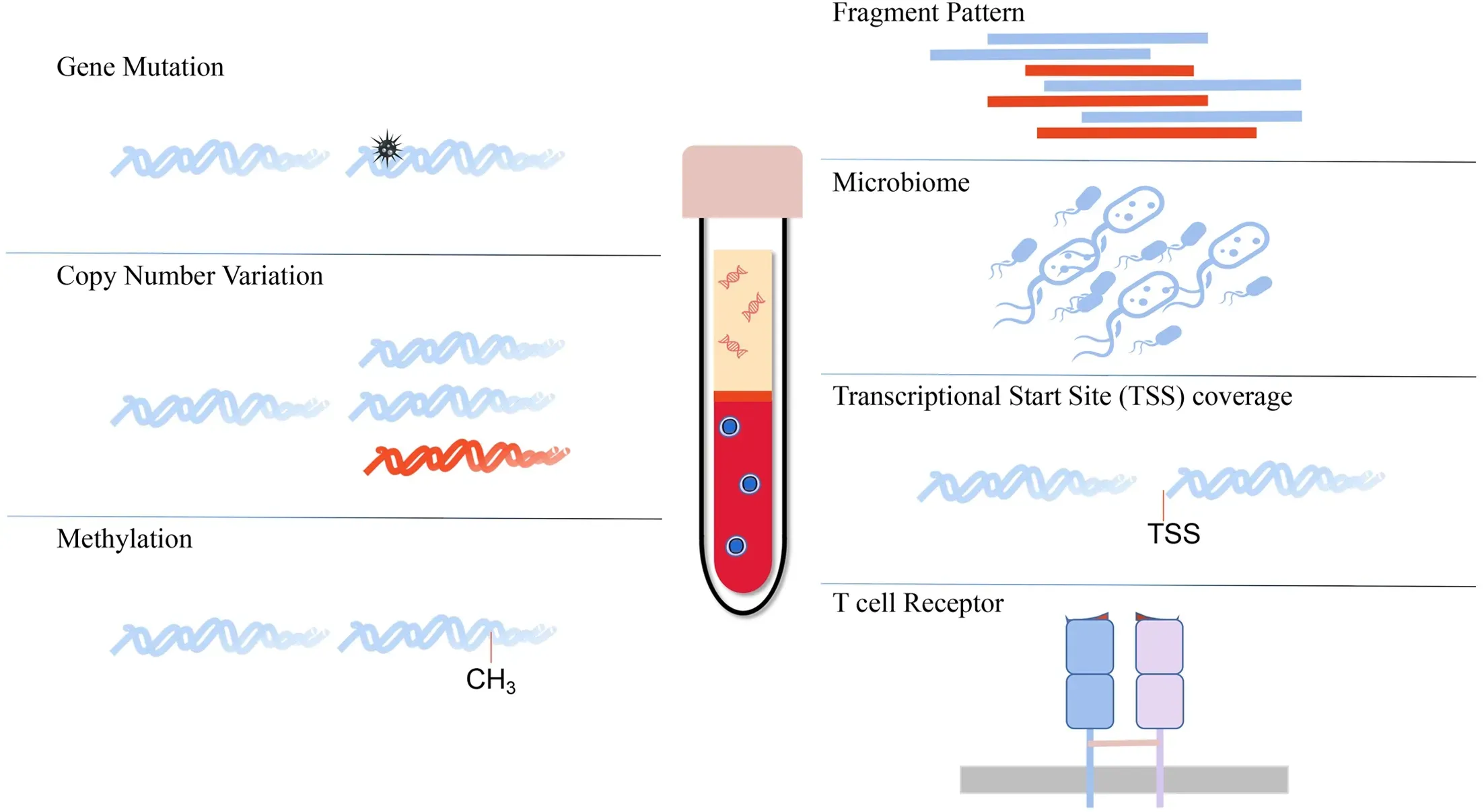
Figure 1 Diverse plasma cfDNA analysis techniques.cfDNA,cell-free DNA;TSS,transcriptional start site.
Canonical cfDNA analysis methods
Testing cfDNA mutations,fusions and copy number alterations
Tumor genotyping (mutations,fusions and copy number alterations) to identify oncogenic driver mutations and mechanisms of resistance to targeted therapeutics has become important in precise oncology (28-30).Most commonly used NGS techniques enable the reliable detection and genomic profiling of cfDNA samples with effects comparable to those of tumor biopsy sequencing,particularly among patients with advanced diseases (23,31-36).
Detection of cfDNA mutations,fusions and copy number variations (CNVs) has been widely used in some cancer types for genomic profiling and treatment selection(19,37-44).The National Comprehensive Cancer Network(NCCN) guidelines (version 4.2020) for non-small cell lung cancer (NSCLC) recommend repeated testing forEGFR,ALK,ROS1,BRAF,METandRETthrough biopsy or plasma testing if insufficient tissues are available.For hormone-receptor (HR) positive/human epidermal growth factor receptor 2 (HER2) negative breast cancer,the NCCN guidelines recommend an assessment ofPIK3CAmutations with tumor tissues or liquid biopsies to identify candidates for alpelisib plus fulvestrant treatment.ERBB2(HER2) plasma copy number detection in cfDNA can be used to guide anti-HER2 therapy in patients with colorectal cancer (45).
In addition to treatment selection,key potential applications of recent plasma ctDNA genomic profiling include risk stratification,response assessment and resistance monitoring (44,46-53).Plasma ctDNA levels combined with the gene mutation status have been examined as prognostic biomarkers across multiple cancer types for risk stratification.For example,in a clinical trial of patients withBRAFV600mutation-positive metastatic melanoma treated with dabrafenib or trametinib,patients negative forBRAFmutations in cfDNA had longer progression-free survival (PFS) and overall survival (44).Regarding the response assessment,studies of ctDNA in patients with advanced pancreatic cancer have reported that a decrease in plasma levels of mutantKRAScfDNA two weeks after treatment appears to be an early indicator of the response to chemotherapy (54,55).Plasma ctDNA analysis has also contributed to monitoring resistance to targeted therapies (19,56-58).For instance,NCCN guidelines support plasma-based mutantEGFRT790M testing to identify acquired resistance to EGFR TKI treatment in patients with NSCLC (37).Acquired resistance to osimertinib in patients withEGFR-mutant NSCLC is mediated by various mechanisms,includingMETamplification,HER2 amplification and various fusions(NTRK,RET,ALKandBRAF),which are potentially detectable by plasma ctDNA NGS (38,39).Plasma ctDNAidentified BRCA reversion mutations have been shown to indicate acquired resistance to PARP inhibitor treatment in patients with prostate cancer (59,60).
DNA methylation testing
DNA methylation is a key epigenetic change involving the addition of a methyl group to cytosine nucleotides,and this modification is used to control genes and their genetic programs (61-63).Epigenetic reprogramming plays an important role in carcinogenesis.The unique levels and patterns of cytosine methylation reflect the tissues of origin and the timing that epigenetic reprogramming has occurred (63).Most types of cancers exhibit a DNA methylation landscape involving the net loss of global DNA methylation and an increase in the levels of methylcytidines at regulatory regions.Thus,this methylation landscape may serve as a potential cancer biomarker to identify the cancer type and stage (64).
A previous study enrolling 6,689 participants (2,482 with cancer,4,207 without cancer) indicated that cfDNA sequencing leveraging informative methylation patterns detected more than 50 cancer types across all stages with high specificity (65).In another study,a targeted set combining genomic alterations (TP53,RB1,CYLDandAR)and epigenomic alterations (hypomethylation and hypermethylation of 20 differentially methylated sites)applied to ctDNA was capable of identifying patients with neuroendocrine prostate cancer (56).Detection of earlystage tumors is still difficult due to the limited amount of ctDNA released into circulation.A methodology termed cell-free methylated DNA immunoprecipitation and highthroughput sequencing (cfMeDIP-seq) was reported that these methylated cfDNA fragments could describe comprehensive profiling of methylated cfDNA and detect cancer in early stages (66).Two studies successively used this technology for the early detection of renal cell carcinomas and the diagnosis of central nervous system tumors (67,68).
Novel cfDNA analysis methods
Fragmentation pattern detection
The cfDNA fragment length is approximately 167 bp in healthy individuals,suggesting release from apoptotic caspase-dependent cleavage (69,70).As cancer cells usually have an altered chromatin structure and other genomic and epigenomic abnormalities,the lengths of cancer-driven cfDNA fragments are more variable than those of noncancer cfDNA (71,72).Tumor-guided personalized deep sequencing and xenograft experiments were performed to establish the size distribution of mutant cfDNA,and an enrichment of cancer cell-derived cfDNA with fragment sizes ranging from 90 bp to 150 bp was observed,which are shorter than non-cancer cell-derived cfDNA fragments.In particular,cfDNA fragments bearing tumor-specific mutations were significantly shorter than fragments without these mutations (73).As cancer-cell cfDNA fragments exhibit a significant difference in length compared with non-cancer-cell cfDNA fragments,the cfDNA fragmentation pattern may serve as a sensitive biomarker to detect cancer.
An approach called“DNA evaluation of fragments for early interception (DELFI)”was developed to specifically and accurately detect a large number of abnormalities in cfDNA by performing genome-wide analysis of fragmentation patterns (25).The first step is to remove low-quality reads and irrelevant reads to obtain highquality sequencing reads.In particular,duplicated reads,low mappable reads and blacklist region reads were removed.Additionally,the length of genome bins is fixed to optimize fragmentation patterns.Whole genome autosomes were divided into three forms,including isometric,adjacent and nonoverlapping bins,with lengths ranging from tens of kb to several Mb,and the number of reads within different intervals was counted.Subsequently,a locally weighted scatterplot smoothing (LOWESS)regression analysis was applied to calculate the guanine and cytosine (GC)-adjusted coverage and account for biases in coverage attributable to GC content.Finally,researchers calculated the ratios of the short to long fragmentation profile for each individual and compared the ratios in the two groups using a Wilcoxon rank sum test to compare the variability of fragment lengths from two groups.
The cfDNA fragmentation pattern can be detected as a proof-of-principle approach in tumor diagnosis.Sensitive detection of genomic alterations in plasma cfDNA relies on the amount of ctDNA released by tumor cells.Notably,low-pass genome sequencing of cfDNA sensitively discovers tens to hundreds of tumor-specific abnormalities through a cfDNA fragmentation pattern analyses,while high-depth genome sequencing is needed to detect tumorderived alterations in a cfDNA mutation analysis.As the cfDNA fragmentation pattern reflects a cell-type specific nucleosome occupation pattern,this detection method is also useful to identify the tissue of origin.Cristianoet al.analyzed the fragmentation patterns of 245 healthy individuals and 236 patients with various types of cancer,including breast,colorectal,lung,ovarian,pancreatic,gastric and bile duct cancer (25).The authors developed a machine learning model incorporating fragmentation patterns,which had sensitivities of detection ranging from 57% to >99% among the seven cancer types at 98%specificity,with an overall area under the curve (AUC) of 0.94.Furthermore,this approach can be combined with mutation-based cfDNA analyses to identify the tissue of origin in 91% of patients with cancer.Mouliereet al.surveyed cfDNA fragment sizes in 344 plasma samples from 200 patients with 18 different cancer types and 65 healthy controls (73).They integrated fragment length and copy number analyses of cfDNA to achieve an AUC>0.99 compared to an AUC<0.8 without fragmentation features in advanced cancer identification.More specifically,increased identification of cfDNA from patients with glioma and renal and pancreatic cancer was achieved with an AUC>0.91 compared to an AUC<0.5 without fragmentation features.
Transcriptional start site (TSS) coverage
TSS is the location where transcription starts at the 5’-end of a gene sequence.Its accessibility,which is affected by nucleosome occupancy,is associated with gene activation or silencing in a tissue-specific manner (74,75).Wholegenome sequencing of cfDNA and identification of TSS coverage can provide functional information about cells releasing their DNA into circulation.Transiently,it will show depleted coverage at the TSS for active genes.In contrast,at promoters of inactive genes,increased coverage may reflect the denser nucleosome packaging of repressed genes (76,77).
Based on the whole genome sequencing of cfDNA,Ulzet al.established a method for analyzing TSS coverage to predict gene expression in specific tissues (5).The first step is to locate TSSs in the reference genome by searching the Ensembl database.After removing low-quality reads,the sequences were aligned to obtain BAM files and subsequently identify coverage around TSS locations.Next,the authors identified nucleosome-depleted regions(NDRs) as open chromatin regions,which were defined as from -150 bp to +50 bp around the TSS.Then,NDR coverage was normalized to the mean coverage of surrounding regions:TSS coverage from -3,000 bp to-1,000 bp and from +1,000 bp to +3,000 bp.Finally,normalized NDR coverage was used to predict gene expression activity.For one specific gene in bulk samples,if the normalized NDR coverage in most of the samples is less than 1,this gene is predicted to be active.If the majority of values is greater than 1,this gene is predicted to be silent.
Due to the TSS coverage of cfDNA possesses sensitivity and accuracy to predict whether genes are expressed,it can be used as an informative tool to determine the expression of cancer-related genes in primary tumors from blood samples.This information may be used in disease stratification for treatment decisions.For example,Ulzet al.first performed RNA-seq of matched primary tumors in addition to whole-genome sequencing of cfDNA in proof-of-concept studies (5).They obtained the 100 most highly expressed genes from RNA-seq analysis of the primary tumor and found that >85% were correctly classified in the expressed cluster by the TSS coverage analysis.This approach was suitable for analyzing expression levels of specific single genes,which may serve as biomarkers for tumor treatment.The authors analyzed 426 plasma samples from patients with metastatic cancer(colon,128;prostate,139;breast,125;lung,31;other tumor entities,3) to test whether this approach is broadly applicable.They found that 51.6% of these samples had at least 100 genomic bins suitable for the TSS coverage analysis.Specifically,certain regions,such as high-level amplifications,which frequently contain cancer driver genes,were always amenable to these analyses.
T cell receptor sequencing
Immune checkpoint inhibitors (ICIs) enhance antitumor immune responses by restoring T cell function (78,79).The identification of indicators of the response to immunotherapy is key for treatment decisions (80).Most researchers have focused on identifying tumor cell states(81),while recent studies report that infiltrated immune cell types and states are changed in response to immunotherapy.Ribaset al.characterized 102 tumor biopsies obtained from 53 patients with metastatic melanoma treated with the PD-1 antibody pembrolizumab.PD-1 blockade increases the frequency of T cells,B cells and myeloid-derived suppressor cells in tumors,while CD8+effector memory T-cells were the main expanded Tcell phenotype detected in patients in response to therapy(82).Riazet al.reported reduced mutation and neoantigen loads in patients with drug-responsive advanced melanoma after treatment with the anti-PD1 antibody nivolumab.Moreover,transcriptomic results showed increased numbers of CD8+T cells and natural killer cells that correlated with the treatment response.T cell receptor sequencing (TCR-seq) showed that expanded T cell clones were accompanied by neoantigen loss (83).Huanget al.identified pharmacodynamic changes in circulating exhausted CD8 T cells (Tex cells) after treatment with the PD-1-targeting antibody pembrolizumab (84).In addition,two studies identified correlations between T cell repertoires and CD8+memory effector cytotoxic T cells in peripheral blood with the response to ICIs in patients with metastatic melanoma and may serve as dynamic biomarkers of immune activation (26,85).
T cell maturation occurs along with clonal reduction and substantial T-cell death because progenitor cells must undergo rounds of selection before they become immunocompetent naïve T cells (86).As dying cells release DNA into the circulation,T cell-derived cfDNA can be sequenced.Complementarity determining region-3 (CDR 3) of the TCR,which is highly variable,is unique to individual T cell clones (87,88).Sequencing CDR3 regions in cfDNA may provide methods to monitor T cell states.
Recently,Valpioneet al.performed a TCR-seq analysis of peripheral blood mononuclear cells (PBMCs) with paired cfDNA to assess early immune activation following ICI treatment (26).The rearrangement efficiency score(RES) [productive/(productive+nonproductive)] was directly used to assess the TCR region CDR3 as a measure of TCR changes in PBMCs and cfDNA.In healthy donors,the level of nonproductive TCR sequences in cfDNA was higher than that in PBMCs,suggesting that nonproductive TCR sequences were released by T cells from a failure of thymic selection.After initial ICI treatment,the cfDNA RES was higher in patients who subsequently responded to ICIs,while the PBMC RES in both responders and nonresponders was 0.A higher cfDNA RES indicated increased peripheral T cell turnover in responding patients.Moreover,flow cytometry results revealed that the change in cfDNA RES was caused by the expansion of a subset of immune effector T cells.
Based on this finding,the activation of this immune effector T cell population may be applied to monitor the early immunotherapy response and thus may guide the next step of treatment selection (89,90).Immunological changes are induced by multiple factors.Future studies should focus on combining more biomarkers in serial TCR-seq analyses of cfDNA to achieve high accuracy and specificity and to translate the available techniques into clinical use.
Microbiome cfDNA analysis
In the past few years,studies have indicated that the microbiome participates in modulating cancer initiation,progression and metastasis,as well as the response to cancer therapy (91-98).For example,Fusobacteriumand its associated microbiome colonize both primary and metastatic sites of human colorectal cancers.Treatment of mice bearing xenografts with the antibiotic metronidazole reduced the bacterial load,cancer cell proliferation and overall tumor growth (91).The microbiota was also discovered to play a role in mediating tumor resistance to the chemotherapeutic drug gemcitabine in colon carcinoma models (93,94). The local microbiota provokes inflammation associated with lung adenocarcinoma progression by activating lung-resident γδ T cells (95).By examining the oral and gut microbiomes of patients with melanoma undergoing anti-PD-1 immunotherapy,Gopalakrishnanet al.observed significant differences in the diversity and composition of the gut microbiome in responders compared with non-responders (99).Characterization of the microbiome in patients with multiple tumor types indicated distinct microbial compositions in these patients (27).For example,theFirmicutesandBacteroidetesphyla were the most abundant species detected in patients with colorectal tumors,whileProteobacteriadominated the microbiome of patients with pancreatic cancer.The microbiomes of patients with breast,lung,and ovarian cancer also showed distinct tumor type-specific compositions (100-102).
Based on accumulating evidence,blood-based microbial DNA (mbDNA) is clinically informative in cancer(103,104).However,due to the low microbial biomass,problems of contaminants and batch effects hampered the use of blood-based microbial DNA detection in the clinic.Pooreet al.established a pipeline to analyze mbDNA in blood,which used improved algorithms for eliminating contaminant sequences and machine learning to identify microbial signatures (27).First,whole genome sequencing is employed in mbDNA profiling.After removing the irrelevant read pairs that map to the human reference genome,the remaining reads are mapped to known bacterial,archaeal and viral genomes with the ultrafast Kraken algorithm or Shogun algorithm in RepoPhlan.This database contains 5,503 viral genomes and 66,279 bacterial or archaeal genomes.Next,the batch effect of datasets are corrected using normalization methods,such as the Voom algorithm.Furthermore,the authors employed machine learning methods to identify microbial signatures that discriminate among various types of cancer and compared their performance.
By implementing this pipeline,Pooreet al.analyzed 18,116 tumor samples from 10,481 patients with 33 different tumor types in The Cancer Genome Atlas(TCGA) database,together with nonneoplastic tumoradjacent tissues and blood samples,as well as matched tissues from individuals without cancer (27).By reanalyzing whole-genome sequences as well as RNA-sequencing data from TCGA,the authors successfully established microbial signatures to distinguish tumor and nontumor tissues and to identify tumor types.Next,they validated their data by analyzing cell-free plasma samples,including samples from 69 individuals without cancer and 100 patients with prostate,lung or skin cancer.The accuracy of cfDNAbased detection was similar to that of the tumor biopsy analysis and tumor type identifications.
Blood-based microbial DNA analysis has great potential for tumor detection and tumor type identification,even low-grade tumor stages,with a high discriminatory rate among healthy individuals and patients with cancer.However,more investigations must be performed to address technical and biological factors limiting the analysis of cancer sequencing data for microorganisms with a low biomass.
Summary
Novel techniques in cfDNA analysis,including fragmentation patterns,TSS coverage,TCR changes and microbial signatures,have wide clinical applications(Figure 2).These novel techniques reflect the status of epigenetic modification,the immune microenvironment and the microbiome in tumor tissues,which play important roles in carcinogenesis.Thus,a deep understanding of the related carcinogenesis mechanism is required,which will provide more biomarkers to test.For example,key transcription factors and related genes involved in tumor initiation can be used as targets in the TSS analysis.Further validation in more patients with different types of cancer is necessary to effectively translate these novel techniques into clinical applications. Moreover,the combination of various methods of cfDNA analysis will add value to future use and increase the specificity and sensitivity.
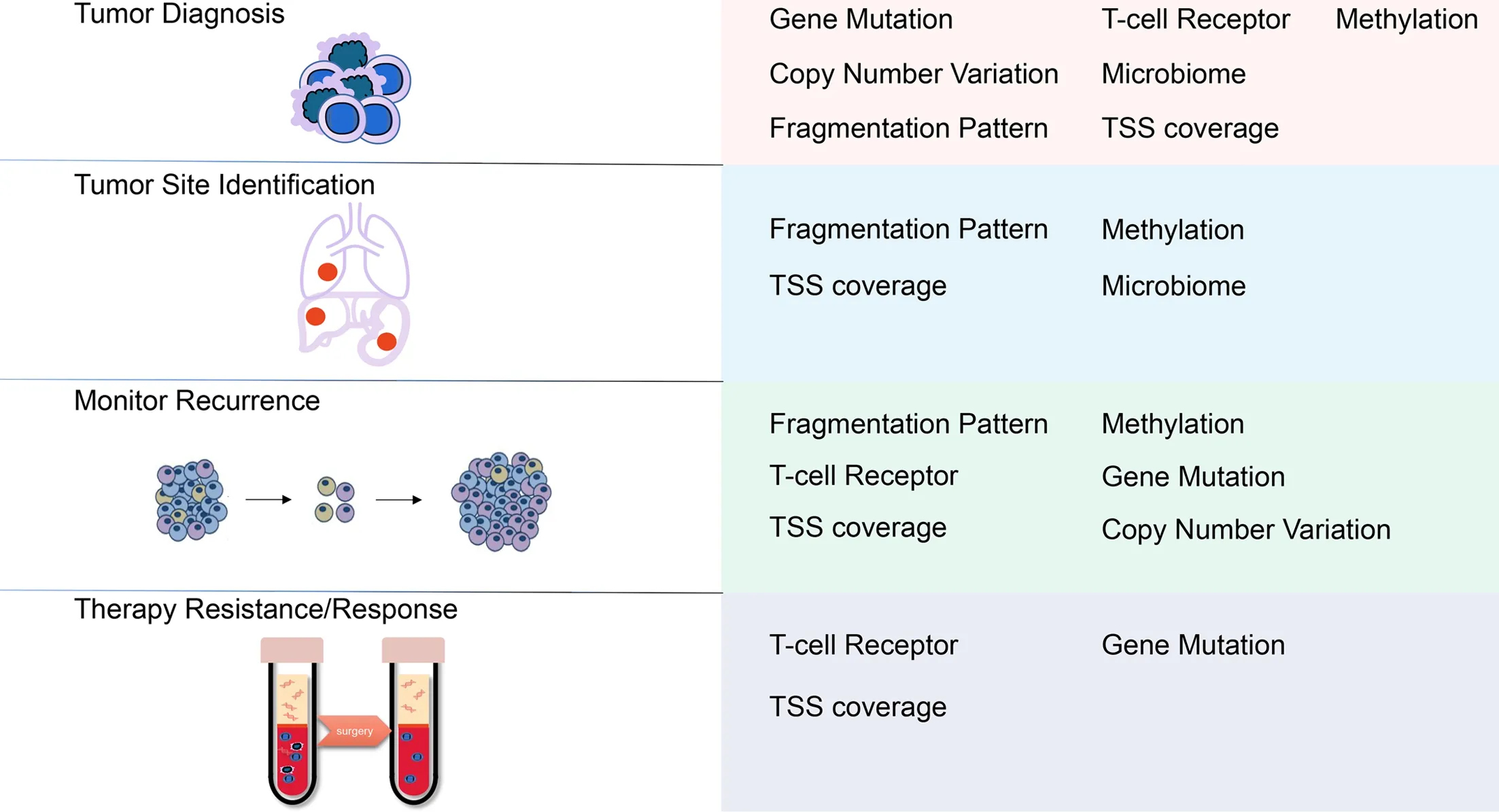
Figure 2 Potential clinical applications of cfDNA technologies.cfDNA,cell-free DNA;TSS,transcriptional start site.
Acknowledgements
This study was supported by the Beijing Natural Science Foundation (No.Z190022) and the National Natural Science Foundation of China (No.81972680,81773292 and 82072748).
Footnote
Conflicts of Interest:The authors have no conflicts of interest to declare.
杂志排行

Chinese Journal of Cancer Research的其它文章
- Prudent application of single-cell RNA sequencing in understanding cellular features and functional phenotypes in cancer studies
- ZNF292 suppresses proliferation of ESCC cells through ZNF292/SKP2/P27 signaling axis
- Development and validation of a questionnaire-based risk scoring system to identify individuals at high risk for gastric cancer inChinese populations
- Associations of centralization with health care quality for gastric cancer patients receiving gastrectomy in China
- Identification of lymph node metastasis by computed tomography in early gastric cancer
- Deep learning model improves radiologists’ performance in detection and classification of breast lesions