基于密度泛函理论从分子层面分析原油中镍/钒卟啉化合物
2022-01-14杨清河
浦 宁,张 文,黄 健,杨清河,徐 超
(1.中国石化 石油化工科学研究院,北京 100083;2.清华大学 核能与新能源技术研究院,北京 100084)
石油是人类社会消费的主要能源之一。国际能源机构(IEA)近几年数据显示,2018年全球原油消费4.5×106ktoe,在能源消费中占比为31.5%,居于各类能源消费量之首;2019年和2020年原油消费分别为99.7 Mbbl/d和91.0 Mbbl/d(全年分别约5.09×106ktoe和4.65 ×106ktoe)[1-2]。近年来,中国经济的高速发展使得能源需求日益扩大,国家统计局官网数据表明,2000—2019年,中国年石油消费量攀升184.1%;相比之下,中国原油年开采量基本维持恒定,在3×105ktoe附近徘徊[3]。目前中国原油对外依存度高达70%,这对中国能源安全存在一定的威胁。全球原油的供需错位、地缘政治等诸多因素导致油价波动,使得国际原油市场上一些廉价劣质原油以及渣油具备了利用价值。劣质原油价格低、加工利润空间较大,但处理困难。另一方面,充分利用劣质原油也是弥补石油资源短缺的重要途径[4]。
原油中的金属元素对炼油工艺流程影响巨大,并且主要存在于渣油中。这些金属元素除了造成换热管道结垢外,Ni、V等重金属元素还会造成催化剂中毒[5-7],影响催化剂寿命。因此,脱除原油中的金属意义重大。Na、Ca、Mg等可溶性无机盐离子通常存在于乳化原油的水相中,通过电脱盐工艺可以对其实现脱除[8]。但Ni、V等过渡金属离子通常以金属-卟啉化合物的形态溶解于油相[9-10],脱除相对困难[11]。目前可用于脱除Ni/V卟啉化合物的方法主要有加氢处理法、溶剂抽提法、螯合分离法等[12-16]。因此,从分子层面研究原油金属卟啉结构性质对于原油脱Ni/V具有基础性的指导意义。
密度泛函理论在金属-有机配体相互作用研究中有着广泛的应用[17-19],在卟啉络合物分子尺度的研究中也发挥着重要作用[20]。Calborean等[21]在PBE/TZP水平上研究了BCP8桥卟啉和Ni2+的相互作用,考察了配合物的构象、二聚体的形成等问题。Wang等[22]使用B3LYP杂化泛函,对V、Ni、Co、Cu使用LANL2DZ基组,配体原子使用6-31++G(d,p)基组,研究了V、Ni、Co、Cu和八乙基卟啉形成的配合物的结构参数、电荷分布、WBI键级(Wiberg Bond Index)、紫外光谱等性质。计算结果与实验规律吻合良好,表明在B3LYP/LanL2DZ/6-31++G(d,p)理论水平上可以很好地模拟V、Ni、Co、Cu卟啉配合物的性质。Stoyanov等[23]比较了14种泛函对镍卟啉及钒卟啉的计算效果,表明PBE和PW91泛函优化镍卟啉及钒卟啉的几何结构、计算金属-卟啉环结合能效果良好。Roy等[24]在B3LYP/LanL2DZ理论水平下模拟了Co-卟啉配合物的几何结构、电离势、电子亲合能、结合能、红外光谱、拉曼光谱等性质。Herritsch等[25]在PBE/def2-TZVPP理论水平下对镍卟啉及镍咔唑的几何结构进行优化,并使用PBE/TZ2P研究了不同镍络合物的电子特性、Ni—N键长及结合能。
文献[26-35]表明,原油中存在的Ni/V卟啉化合物主要包括ETIO型、DPEP型、DI-DPEP型、RHODO-ETIO型、RHODO-DPEP型、RHODO-DI-DPEP型等。笔者基于密度泛函理论系统研究了上述卟啉环和Ni2+、VO2+离子的相互作用,从能量角度分析了金属-配体间的相互作用机制;研究了上述金属卟啉配合物的几何结构、配位模式以及卟啉分子取代基变化对配位特性的影响;并从理论层面上分析了石油卟啉分子层面的化学性质,对深入理解金属-卟啉相互作用,解释原油或渣油脱金属的实验规律提供了理论支撑。笔者从基础配位化学角度,通过量子化学计算,从分子层面探究了原油中的Ni2+/VO2+-卟啉配合物性质,对于原油脱Ni/V技术的研究具有理论指导作用。再者,国内外学者提出“分子炼油”、“组分炼油”的概念,即按照石油构成分子的类别进行石油炼制,从而极大程度挖掘石油的原料价值[36-40]。因此,笔者从分子层面对原油中关键分子进行系统研究,对炼油行业从目前依据馏程切割原油向组分炼油甚至分子炼油发展有着理论支撑作用。
1 计算方法
卟啉结构的基础是卟吩核,当吡咯环上的氢原子被取代基取代,即形成卟啉配体。笔者选择最具代表性的石油卟啉分子进行理论研究:ETIO型、DPEP型、DI-DPEP型、RHODO-ETIO型、RHODO-DPEP型、RHODO-DI-DPEP型6种类型,对应的代表性分子结构式如图1所示。通过密度泛函理论(DFT)对各卟啉分子、脱质子后形成的卟啉阴离子、卟啉-金属配合物进行几何结构优化,进一步计算了各种金属-卟啉配合物解离成金属离子和卟啉阴离子的解离能,并从分子几何结构、电荷分布、前线轨道等多角度对Ni2+/VO2+-卟啉体系进行了分析。C、H、O、N原子使用6-31G(d,p)基组;Ni、V金属离子使用LanL2DZ或SDD2种赝势基组分别进行计算,并将计算结果进行对比。泛函选择B3LYP。计算使用Gaussian09软件,所有的计算均在清华高性能计算平台(Center of high performance computing,Tsinghua University)完成[41]。
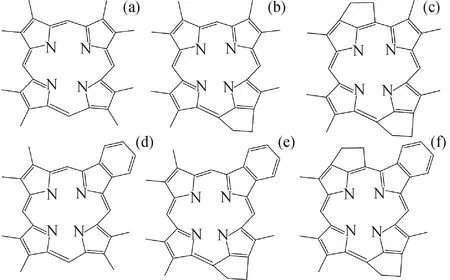
图1 原油中代表性的卟啉分子结构Fig.1 Typical porphyrin molecule structures in petroleum(a)ETIO;(b)DPEP;(c)DI-DPEP;(d)RHODO-ETIO;(e)RHODO-DPEP;(f)RHODO-DI-DPEP
2 结果与讨论
2.1 卟啉离子的结构性质分析
卟啉是由卟吩核在吡咯环上引入取代基而形成的一类化合物,卟啉分子可以脱除2个氢离子,而形成卟啉阴离子,DFT优化的卟啉离子结构如图2所示。图2中,从空间角度分析:卟啉离子呈现平面结构,4个吡咯环的N原子朝向环内侧,可以实现对金属离子的四齿螯合;卟啉离子呈现出良好的利于配位的预组织结构,配位N原子形成的空腔中心到N原子中心距离约0.2 nm,与Ni2+、VO2+的离子尺寸比较匹配,空间上有利于络合[42-43]。
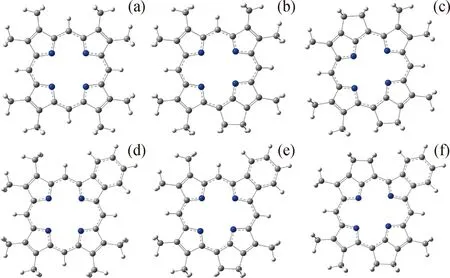
Blue ball—N atom;Grey ball—C atom;White ball—H atom图2 原油中主要卟啉化合物的卟啉离子结构Fig.2 Structure of porphyrin anions of dominant porphyrin compounds in petroleum(a)ETIO;(b)DPEP;(c)DI-DPEP;(d)RHODO-ETIO;(e)RHODO-DPEP;(f)RHODO-DI-DPEP
从电荷角度分析,DFT计算得到笔者所研究的6种卟啉阴离子配位N原子上的平均Mulliken电荷基本一致,为-0.523~-0.524 e;以ETIO卟啉离子为例,绘制由DFT计算得到的静电势图(Electrostatic potential,ESP)如图3(a)所示,图中红色代表负电荷,卟啉环空腔中心存在密集的负电荷,因此有利于螯合金属离子。从分子轨道角度分析,图3(b)展示了ETIO卟啉离子的最高占据轨道(Highest occupied molecular orbital,HOMO)。卟啉环呈现平面结构,4个吡咯环的α-C原子通过 4个次甲基连接,环上C/N原子的p轨道垂直于卟啉环平面,表现出显著的共轭作用。因此卟啉环结构稳定,且共轭的π轨道可以与金属(Ni或V)的3 d 轨道形成显著的轨道重叠。总地来说,空间、电子、轨道方面均有利于卟啉离子和Ni2+/VO2+形成稳定的配合物。因此,原油中Ni2+/VO2+-卟啉配合物结构稳定,也较难脱除。

Blue ball—N atom;Grey ball—C atom;White ball—H atom;Red represents negative charge图3 ETIO离子的静电势图(ESP)和最高占据轨道HOMO轨道Fig.3 Electrostatic potential (ESP)diagram and the highest occupied molecular (HOMO)orbital of the ETIO anion(a)ESP diagram;(b)HOMO orbital
2.2 Ni2+/VO2+-卟啉配合物的结构性质分析
几何优化时,对于配合物的配位核心金属离子尝试使用2种基组,分别为LanL2DZ基组或更大的SDD基组。卟啉环通过4个N原子与金属离子配位,4个Ni—N配位键的平均键长列于表1中,优化的Ni2+-卟啉配合物结构如图4所示。由表1可知,在LanL2DZ或SDD 2种基组下,配位键键长差异仅仅在0.001 nm以内,表明Ni2+/VO2+-卟啉配合物的几何结构优化对基组的敏感性较小。随卟啉环取代基的改变,配位键键长变化较小,当卟啉环上的环烷基取代基结构增加(ETIP→DPEP→DI-DPEP,RHODO-ETIP→RHODO-DPEP→RHODO-DI-DPEP)时,Ni—N配位键键长缩短;当卟啉环上的取代基由甲基变为苯基(ETIP→RHODO-ETIP,DPEP→RHODO-DPEP,DI-DPEP→RHODO-DI-DPEP)时,Ni—N配位键键长增长。这一规律同样出现在VO2+-卟啉配合物中(下文讨论)。这是由于环烷基具有供电子作用,促进配体和金属配位,使得石油卟啉配合物配位键键长缩短;而苯基具有吸电子特性,不利于配体和金属配位,使得配位键键长增长。

表1 Ni2+/VO2+-卟啉配合物中关键键长Table 1 Key bond length of Ni2+/VO2+-porphyrin complexes
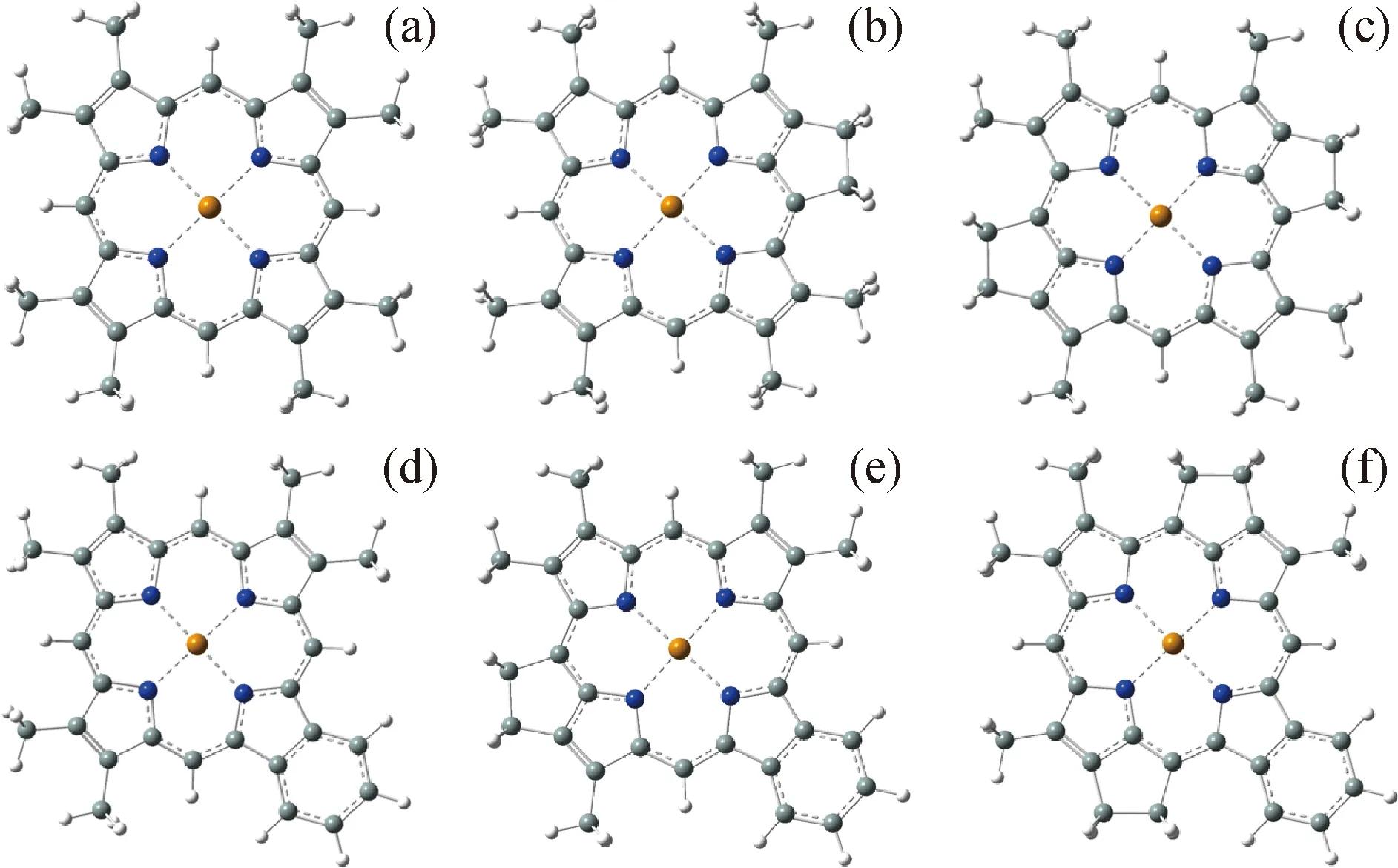
Blue ball—N atom;Grey ball—C atom;White ball—H atom;Yellow ball—Ni atom图4 Ni2+-卟啉配合物的结构Fig.4 Structure of Ni2+-porphyrin complexes(a)ETIO;(b)DPEP;(c)DI-DPEP;(d)RHODO-ETIO;(e)RHODO-DPEP;(f)RHODO-DI-DPEP
优化后的VO2+-卟啉配合物结构如图5所示,卟啉环同样是通过4个N原子和V直接配位。4个V—N配位键的平均键长及V=O键长列于表1中,自由的VO2+离子中,V=O键长为0.1518 nm(LanL2DZ)/0.1498 nm(SDD)。当VO2+离子和卟啉离子络合后,V=O键长出现了显著的增长,可见卟啉离子和轴向O原子存在对V的配位竞争,卟啉离子的配位削弱了V=O之间的配位作用。随着卟啉环取代基的改变,配位键键长变化较小。与Ni2+-卟啉配合物配位键键长随卟啉取代基变化的趋势一致:当卟啉环上的取代基由烷基变为环烷基(ETIP→DPEP→DI-DPEP)时,V—N配位键键长缩短;而当吡咯环苯并以后(形成RHODO-ETIO),配位键键长增长;在含有苯并吡咯结构的配合物中,随着环烷基的引入,配位键键长也出现缩短的趋势(RHODO-ETIP→ RHODO-DPEP→RHODO-DI-DPEP)。

Blue ball—N atom;Grey ball—C atom;White ball—H atom;Green ball—V atom;Red ball—O atom图5 VO2+-卟啉配合物的结构Fig.5 Structure of VO2+-porphyrin complexes(a)ETIO;(b)DPEP;(c)DI-DPEP;(d)RHODO-ETIO;(e)RHODO-DPEP;(f)RHODO-DI-DPEP
比较VO2+-卟啉和Ni2+-卟啉配合物结构,如图6所示。可以发现,VO2+-卟啉和Ni2+-卟啉配合物结构的显著差异为Ni2+-卟啉配合物中的Ni原子和卟啉环共面,VO2+-卟啉配合物中的V原子和卟啉环不共面;这主要是由于V的离子半径稍大,空间位阻效应使得V位于卟啉环中稍微偏离卟啉环平面的位置,同时O原子与卟啉环对V原子存在竞争配位。
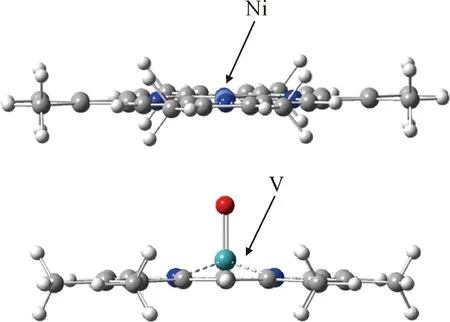
图6 Ni2+/VO2+-ETIO配合物结构对比Fig.6 Structure comparison of Ni2+/VO2+ complex with ETIO
2.3 Ni2+/VO2+-卟啉配合物的能量分析
为进一步分析卟啉和Ni2+或VO2+离子之间的相互作用强弱以及取代基效应,基于DFT理论,对金属离子选择了2种基组(LanL2DZ或SDD),计算了6种卟啉离子与Ni2+/VO2+配合物的气相解离能,如表2所示。可以发现,卟啉阴离子和Ni2+或VO2+离子之间相互作用强烈,络合十分稳定,因此实际生产中原油脱Ni/V存在明显的技术难度。卟啉环上烷基的改变对解离能影响不大,但随着卟啉环上苯基的引入,解离能显著减小,可见苯基的吸电子效应削弱了金属-卟啉之间的相互作用;这与前文讨论苯基引入使得平均M(金属)—N键长增长是一致的。DFT计算结果表明,钒卟啉的解离能低于镍卟啉,可见相比于Ni2+,VO2+和卟啉离子配位能力要弱一些,这与实验中V的脱除比Ni要容易是一致的[44-45]。除此以外,笔者选择较小基组(LanL2DZ)和较大基组(SDD)计算得到的解离能差异很小(在0.3%以内),且2种基组计算得到的趋势完全一致。因此,在计算原子数不太多的体系时(如研究原油中金属离子和卟啉、环烷酸、噻吩等配位特性),为了获得可信的精度,应当考虑SDD或者更大的基组;如果进一步研究Ni2+/VO2+-卟啉配合物和含有上百个乃至数百个原子的沥青质相互作用,在计算资源有限的条件下可以考虑LanL2DZ基组。

表2 Ni2+/VO2+-卟啉配合物的解离能Table 2 Dissociation energy of Ni2+/VO2+-porphyrin complexes
3 结 论
基于密度泛函理论(DFT)系统研究了主要的石油卟啉和Ni2+/VO2+形成的配合物,从分子层面分析了各种石油卟啉的结构特性及配位模式。卟啉环是具有芳香性的平面结构,通过4个N原子和Ni2+或VO2+配位。卟啉环的空腔尺寸和Ni2+、VO2+的离子尺寸比较匹配,空腔中心存在密集的负电荷,共轭的π轨道可以和金属(Ni或V)的3d轨道形成显著的轨道重叠,均有利于螯合金属离子形成稳定的配合物。V原子相对较大的离子半径导致的空间效应及O原子与卟啉环对V原子的竞争配位,使得相比于VO2+,卟啉环和Ni2+配位更加稳固,这与实际加工过程中Ni更难脱除现象一致。卟啉环的取代基对配位有着明显的影响,如供电子的环烷基促进了卟啉和Ni/V的配位;吸电子的苯基削弱了卟啉和Ni/V的配位。
