Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease
2022-01-12AlejandroRodaGabrielSerraMirLaiaMontoliuGayaLidiaTiesslerSandraVillegas
Alejandro R. Roda, Gabriel Serra-Mir, Laia Montoliu-Gaya, Lidia Tiessler,Sandra Villegas,*
Abstract Alzheimer’s disease is a neurodegenerative disease that accounts for most of the 50-million dementia cases worldwide in 2018. A large amount of evidence supports the amyloid cascade hypothesis, which states that amyloid-beta accumulation triggers tau hyperphosphorylation and aggregation in form of neurofibrillary tangles, and these aggregates lead to inflammation, synaptic impairment, neuronal loss, and thus to cognitive decline and behavioral abnormalities. The poor correlation found between cognitive decline and amyloid plaques, have led the scientific community to question whether amyloid-beta accumulation is actually triggering neurodegeneration in Alzheimer’s disease.The occurrence of tau neurofibrillary tangles better correlates to neuronal loss and clinical symptoms and, although amyloid-beta may initiate the cascade of events, tau impairment is likely the effector molecule of neurodegeneration. Recently, it has been shown that amyloid-beta and tau cooperatively work to impair transcription of genes involved in synaptic function and, more importantly, that downregulation of tau partially reverses transcriptional perturbations. Despite mounting evidence points to an interplay between amyloid-beta and tau, some factors could independently affect both pathologies. Thus, the dual pathway hypothesis, which states that there are common upstream triggers causing both amyloid-beta and tau abnormalities has been proposed. Among others, the immune system seems to be strongly involved in amyloid-beta and tau pathologies. Other factors, as the apolipoprotein E ε4 isoform has been suggested to act as a link between amyloid-beta and tau hyperphosphorylation. Interestingly, amyloid-beta-immunotherapy reduces not only amyloid-beta but also tau levels in animal models and in clinical trials. Likewise, it has been shown that tau-immunotherapy also reduces amyloid-beta levels. Thus, even though amyloid-beta immunotherapy is more advanced than tau-immunotherapy, combined amyloid-beta and tau-directed therapies at early stages of the disease have recently been proposed as a strategy to stop the progression of Alzheimer’s disease.
Key Words: aggregation; Alzheimer; amyloid-beta; dementia; immunotherapy;inflammation; neurodegeneration; tau
Introduction
Most of the 50 million people living with dementia worldwide in 2018 suffered from Alzheimer’s disease (AD). AD is a major cause of disability and dependency among elderly people, so economic impact is huge, with an estimate of US$ 1,000,000 million just for the year 2018 (Alzheimer’s Disease International, 2018). AD is a neurodegenerative disease characterized by memory loss,cognitive impairment, and behavioral and psychological symptoms of dementia. The main histopathological hallmarks of AD include the extracellular accumulation of the amyloid-beta (Aβ) peptide in senile plaques and formation of neurofibrillary tangles (NFTs)by hyperphosphorylated tau protein. Mounting evidence supports the assumption that cerebral Aβ deposition begins decades before AD clinical onset and prior to cortical spreading of tau pathology.However, a complete understanding of the role of Aβ, from loss of proteostasis to synaptic toxicity is still lacking. Similarly, it is accepted that tau hyperphosphorylation plays a critical role in neuron death;however, whether it is cause or consequence of disease development remains unknown. Although controversial, the amyloid cascade hypothesis (ACH) is the only hypothesis covering the multifactorial nature of AD (Karran and De Strooper, 2016; Selkoe and Hardy, 2016).The aim of this review is to discuss new findings on the interplay between Aβ and tau in the context of an updated view of the ACH and to summarize the state of the art in AD clinical trials that are the basis for finding subsequent anti-Aβ and anti-tau combined therapies.
Search Strategy and Selection Criteria
Literature search was performed on PubMed until November 2020. Only papers published in English were considered. The key words/terms were aggregation, Alzheimer, amyloid-beta, cascade hypothesis, dementia, crosstalk, interaction, immunotherapy,inflammation, neurodegeneration, and tau.
Amyloid-beta Peptide as the Alzheimer’s Disease Trigger: the Amyloid Cascade Hypothesis
Amyloid precursor protein (APP) is an ubiquitous single-pass transmembrane protein that contains an extracellular domain, a hydrophobic transmembrane domain, and an intracellular domain(Kang et al., 1987). Under physiological conditions, soluble APP (sAPP)is involved in neurite outgrowth, synaptogenesis, synaptic plasticity,and cell survival through N-methyl-D-aspartate and gammaaminobutyric acid receptors modulation, which maintain intracellular calcium homeostasis (Müller et al., 2017; Rice et al., 2019).
APP can be cleaved by a combination of different secretases complexes, following two pathways (Figure 1A). In the nonamyloidogenic APP processing, the combined action of α- and γ-secretases releases sAPPα, P3 peptide (which corresponds to the Aβ17–40/42sequence) and the APP intracellular domain (De Strooper et al., 1998). The amyloidogenic processing of APP is initiated by β-secretase (or β-site APP-cleaving enzyme 1 (BACE1)), instead of α-secretase so sAPPα is released; next, γ-secretase generates Aβ peptides with different C-terminal residues and APP intracellular domain. This cleavage, at what is known as the β-site, can be also performed by BACE2 (59% identity to BACE1 and similar structural organization), although BACE2 tends to cleavage at the β’-site,rendering an N-truncated, non-amyloidogenic β-peptide (Kimura et al., 2016; Wang et al., 2019). Therefore, cleavage by γ-secretase is the one generating heterogeneity at the C-terminus end of Aβ peptide, producing mainly Aβ1–40and, to a lesser extent, the most amyloidogenic Aβ1–42form (Figure 1B). Aβ1–40tends to accumulate in the vasculature whereas Aβ1–42constitutes the predominant form in amyloid plaques.
Three decades ago, mutations inAPP(Citron et al., 1992) (21q11.2–q21) (Figure 1C), as well as in the catalytic units of γ-secretase genes, presenilin 1 (Sherrington et al., 1995) (PSEN1, 14q24.3) and presenilin 2 (Levy-Lahad et al., 1995) (PSEN2, 1q42.2), were reported to cause familial AD (FAD) (Esquerda-Canals et al., 2017b). These mutations result in Aβ accumulation because of amyloidogenic APP processing enhancing. One of the most remarkable FAD mutations is the Swedish mutation, a double mutation at the β-site (K670/N, M671/L) that enhances APP processing by β-secretase and so generates increased levels of Aβ compared with control individuals.In contrast, Icelandic mutation (A673T), located at the β-site as well,lessens amyloidogenic APP processing and so protects against AD onset (Jonsson et al., 2012). Some other relevant mutations onAPPgene are located on the N-terminal part of the Aβ peptide or close to the γ-secretase site (Esquerda-Canals et al., 2017b).
The variety of mutations onPSENgenes encompasses missense mutations, small deletions and insertions, as well as genomic exon deletions, beingPSEN1mutations the most usual in FAD (Park et al., 2017; Bagyinszky et al., 2018; Giau et al., 2018a, b). Although hundreds of mutations on APP, PSEN1, and PSEN2 have been described, they only account for a small part of early-onset AD cases,attributable when AD onset occurs before 65 years of age. Non-FAD early-onset AD cases are clustered with late-onset AD into what is called the sporadic form of the disease (SAD). Several years ago,it was observed that the serum of non-AD individuals shows autoantibodies against Aβ, which indicates that some amyloidogenic processing also occurs in normal conditions. In addition, there is a differential physiological role of BACE1 (which generates Aβ peptide,as mentioned) and BACE2 (which generates both Aβ peptide and an N-truncated form, as mentioned). BACE1 is indispensable for proper astrogenesis, axonal growth and migration, myelination and remyelination, neuronal excitation, and synaptic plasticity; whereas BACE2 is involved in different processes, as glucose homeostasis and pigmentation (Yan, 2017). Studies using physiological Aβ concentrations (pM) show a role in normal synaptic physiology,enhancing long term potentiation, promoting survival of neurons,and promoting a neurogenic effect on neural progenitor cells. In addition, Aβ is involved in the repair of leaks in the blood-brain barrier, promotes recovery from injury, and acts as an antimicrobial and tumor suppressor peptide (Kent et al., 2020).
In 1992, taking into consideration FAD cases, together with the fact that location of APP gene on chromosome 21 leads Down syndrome individuals to suffer from AD (Macleod et al., 2015) by the time they reach middle to late age, the ACH was postulated (Hardy and Higgins,1992) (Figure 2). ACH states that Aβ peptide deposition triggers tau hyperphosphorylation and aggregation in the form of NFTs, and these aggregates lead to inflammation, synaptic impairment, neuronal loss,and thus to cognitive decline and behavioral abnormalities. However,Aβ overproduction is not the only cause of AD since the reduction in Aβ auto-antibodies featuring SAD points to an impairment in Aβ clearance. In consonance, the most well-known genetic risk factor for developing SAD is the apolipoprotein E (APOE) gene, which encodes apolipoprotein E, and the risk specifically occurs when theAPOEε4allele is present (Roda et al., 2019; Andrews et al., 2020).
The fact that several clinical trials for Aβ-immunotherapy-based drugs have failed make made some researchers to question whether the ACH explains the physiopathology of AD by itself (Herrup, 2015).However, the decision on the accelerated approval of aducanumab(Aduhelm, Biogen) was recently make (https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatmentalzheimers-disease). An accelerated decision means that the drug is shown to have an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit to patients and there remains some uncertainty about the drug’s clinical benefit.
Different aggregation states of Aβ coexist in AD brains, monomers,oligomers, fibrils and amyloid plaques (Figure 3A). Experimental evidence suggests that plaques maintain a dynamic equilibrium with toxic oligomer species (Benilova et al., 2012). Monomeric Aβ is an intrinsically disordered protein; hence,, Aβ does not adopt a single folded shape in aqueous environment but acquires an ensemble of conformations which are prone to aggregation in the form of Aβ oligomers (Ball et al., 2011). Aβ oligomers are the transient intermediate states between monomer and fibril formation. Both Aβ oligomers and fibrils are heterogeneous in structure and size and they are known to be the most toxic Aβ species (Benilova et al.,2012), particularly oligomers. Although solving the structure of the Aβ peptide from human samples has been quite challenging, the 3D-structure of both, Aβ1–40and Aβ1–42fibrils has been solved in the last decade (Lu et al., 2013; Qiang et al., 2017). Aβ1–40fibrils from one single patient shows that Aβ1–40peptide monomers aggregate into multiple of three oligomers (trimers, hexamers, nonamers, and dodecamers), where N-termini (DAEFR, residues 1–5) are exposed to the solvent and hydrophobic C-termini are buried in the trimer core(Figure 3B; pdb 2M4J) (Lu et al., 2013). Because the concentration of Aβ1–40is much higher than Aβ1–42, such multiple of three oligomers are found in most studies with animal models for AD, i.e., in the 3xTg-AD model (Giménez-Llort et al., 2013). The structure of Aβ1–42fibrils shows a quite different structure, with two twisted protofilaments stacked in parallel and thus built of “LS”-shaped dimers, with the N-terminus slightly solvent-exposed and the C-terminus completely buried (Figure 3C; pdb 5OQV) (Gremer et al., 2017). How oligomers promote cell toxicity has not been completely unraveled. However,several molecular mechanisms as activation of metabotropic glutamate receptor 5, Caspase-3 activation, N-methyl-D-aspartate receptor interaction, and α7 nicotinic acetylcholine receptor upregulation have been proposed (Bezprozvanny, 2009).
Although amyloid deposits do not correlate with either disease progression or cognitive decline (Hanseeuw et al., 2019), their spatial and temporal emergence are well described (Braak and Braak, 1991).In a first stage, amyloid deposits occur in the frontal, temporal, and occipital lobes of the isocortex. In a second stage, they spread to all the isocortical association areas, except on primary sensory areas and motor field. Disperse amyloid deposits also occur in the frontal and parietal lobes. In this stage, the hippocampus is slightly affected.The final stage is characterized by depositions in primary isocortical areas and progressive spreading to the striatum, thalamus and hypothalamus.
Tau and Its Role in Neurodegeneration
As mentioned before, the lack of success in approving any anti-Aβ therapy up to now, as well as the poor correlation found between cognitive decline and the occurrence of amyloid plaques, have led the scientific community to question whether Aβ accumulation is actually triggering neurodegeneration in AD. The occurrence of tau NFTs better relates to neuronal loss and clinical symptoms (Hanseeuw et al., 2019) and, although Aβ may initiate a cascade of events, tau impairment is likely the effector molecule of neurodegeneration.Tau, apart from being a stabilizer of microtubules and being involved in their dynamics, plays a role in several physiological processes including myelination, axonal transport, neurogenesis,motor function, learning and memory, neuronal excitability, glucose metabolism, iron homeostasis, and DNA protection (Kent et al.,2020). It is important to mention that while Aβ accumulation is characteristic of AD, tau pathology also exists in a group of neurodegenerative diseases known as tauopathies, which are different to AD.
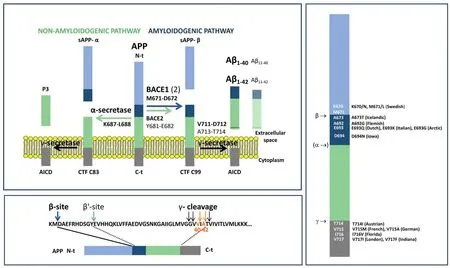
Figure 1 |Amyloid precursor protein (APP)processing.(A) α- and γ-secretase act in the nonamyloidogenic pathway, whereas the activity of β-secretase (or BACE) instead of α-secretase defines the amyloidogenic pathway generating the Aβ peptide. BACE1 hydrolyses the β-site(M671-D672 bond) and BACE2 cleaves the β’-site (681Y-682E bond), rendering an N-t truncated form of the precursor (E11-Xx),although also cleaves the β-site. Thus, β′-site cleavage is amyloidolytic rather than amyloidogenic. Finally, γ-secretase releases the Aβ peptide, or its N-t truncated form,after preferentially cleaving the V711-D712 and A713-T714 bonds rendering Aβ1–40 and Aβ1–42, respectively. (B) Target proteolytic sites in the Aβ peptide. (C) Some of the main FAD-related mutations in human APP. ACID:APP intracellular domain; Aβ: amyloid-beta peptide; BACE: beta-site APP cleaving enzyme;CTF: carboxyterminal fragments; FAD: familial Alzheimer’s disease.
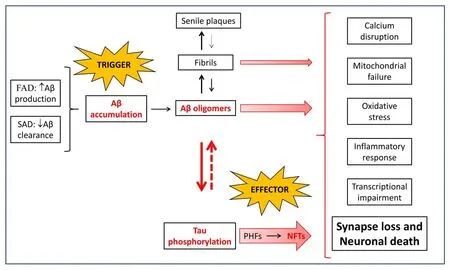
Figure 2 |The amyloid cascade hypothesis (ACH).Whether by an increase in production (FAD) or an impairment in clearance (SAD), Aβ peptide accumulates through an aggregation pathway populated by different species, i.e. monomers,oligomers, fibrils, and amyloid plaques. Aβ oligomers are the most toxic species, and to a lesser extent fibrils, causing calcium disruption, mitochondrial failure, inflammatory response, oxidative stress,transcriptional impairment, synaptic loss and, finally,neuronal death. In addition, Aβ oligomers induce the hyperphosphorylation of tau, which aggregates into oligomers that evolve to paired helical filaments and finally to NFTs. Tau oligomers are also toxic for neurons, with a strong effect on synapses, and,in turn, induce the formation of Aβ oligomers. In addition to this positive feedback between Aβ and tau aggregation, Aβ and tau cooperatively work to lead to neuronal death. Although still controversial, the ACH covers the multifactorial nature of AD. Aβ: Amyloidbeta peptide; FAD: familial Alzheimer’s disease; NFTs:neurofilament tangles; PHF: paired helical filaments;SAD: sporadic Alzheimer’s disease.

Figure 3 |Amyloid fibril formation and threedimensional structure.(A) Aggregation of Aβ from the formation of different kind of oligomers to amyloid fibrils. (B) Aβ1–40 amyloid fibrils are composed by multiple ordered trimers. The N-termini are completely solvent-exposed, whereas the C-termini are buried in the hydrophobic core(PDB 2M4J) (Lu et al., 2013). Each color represents a monomer. (C) Aβ1–42 amyloid fibrils are made of dimers. The N-termini are slightly solvent-exposed,whereas the C-termini are completely buried in the interface between monomers (PDB 5OQV) (Gremer et al., 2017). Each monomer in the dimer shows higher β-content than in the trimer. Each color represents a monomer. Images were generated with the Swiss-PdbViewer software (Guex and Peitsch, 1997). Aβ:Amyloid-beta peptide; PDB: protein data bank.
The link between tau dysfunction and neurodegeneration was set through human genetics. Mutations inMAPT, the microtubuleassociated protein Tau gene, give rise to an inherited form of Frontotemporal Dementia (FTD) and parkinsonism with abundant filamentous tau deposits in the brain, in the absence of Aβ deposits(Goedert, 2015). Six different isoforms of tau are expressed in the adult human central nervous system via alternative splicing of theMAPTgene, which comprises 16 exons and is found on chromosome 17q21.3 (Figure 4A). Regulated expression of exons 2 and 3 gives rise to tau isoforms with 0, 1, or 2 N-terminal inserts, whereas exclusion or inclusion of exon 10 leads to expression of tau isoforms with three(3R) or four (4R) microtubule-binding repeats (Connell et al., 2005).3R or 4R isoforms are predominant in tau inclusions depending on the neurodegenerative disease. Progressive supranuclear palsy (PSP),corticobasal degeneration, argyrophylic grain disease, among others,exhibit overexpression of 4R isoforms; whereas Pick’s disease, and familial FTD and parkinsonism (MAPTmutations such as G272V and Q336R) are mainly characterized by tau inclusions rich in 3R tau isoforms (Irwin, 2016). In other diseases such as AD, Down syndrome or Chronic traumatic encephalopathy, both 3R and 4R isoforms are found in tau tangles. The N-terminal phosphatase-activating domain in tau, which triggers a signaling pathway that disrupts axonal transport, is exposed in all the isoforms. The extent of phosphataseactivating domain exposure and oligomerization is larger for tau aggregates composed of 4R isoforms than 3R isoforms. However,aggregates of all isoforms exhibit enough phosphatase-activating domain exposure to impair axonal transport (Cox et al., 2016).
Tau is predominantly expressed in the central and peripheral nervous system, where it is most abundant in nerve cell axons. In the brain,tau is subject to several posttranslational modifications, including phosphorylation, acetylation, methylation, glycation, isomerization,O-GlcNAcetylation, nitration, sumoylation, ubiquitination, and truncation (Morris et al., 2015), but the role all these modifications play in tau function remains unknown. The most studied tau posttranslational modification is phosphorylation, as despite the significant heterogeneity that exists between and within the different tauopathies, the deposited tau in pathological lesions is invariably highly phosphorylated (Noble et al., 2013). Phosphorylation of tau negatively regulates its ability to interact with microtubules, but whether phosphorylation is a trigger for aggregation remains to be proved. Although aggregated tau is heavily phosphorylated in the human brain, not all phosphorylated tau is aggregated (Goedert et al., 2017). Full-length tau assembles into filaments through its repeats, with the N-terminal half and the C-terminus forming the fuzzy coat (Figure 4B; pdb 6HRE) (Falcon et al., 2018). The filament core contains the repeat sequences that are required to the binding of soluble tau to microtubules, suggesting that physiological function and pathological assembly are mutually exclusive (loss of function).Brains from different individuals with the same tauopathy show the same kind of tau filaments (Falcon et al., 2018), whereas each disease is characterized by its own unique tau fold (Scheres et al.,2020). Cross-seeding experiments have shown that Aβ oligomers induce tau oligomerizationin vitro(Lasagna-Reeves et al., 2010),indicating that early abnormal Aβ oligomerization precedes tau aggregation. Interestingly, it has been reported that structurebased inhibitors of Aβ are able to avoid tau aggregation, suggesting that there is a common aggregation interface in both tau and Aβ(Griner et al., 2019). Although tau is mainly an intracellular protein,extracellular tau is present in the brain and interstitial fluid (Yamada et al., 2011), and it is thought to be secreted under physiological conditions. Microglial cells may propagate tau through exosomedependent mechanisms (Asai et al., 2015). Spread of tau aggregated assemblies may seed and promote aggregation (Falcon et al., 2015)along different brain regions. In AD, deposition of tau aggregates follows a highly stereotyped pattern, beginning in the entorhinal cortex and hippocampus before propagation to other regions. Their stereotypical appearance in nerve cells underlies Braak´s stages of AD (Jellinger, 1998), according to which inclusions form first in subcortical regions, transentorhinal cortex, and entorhinal cortex(stages I and II). They then appear in the hippocampal formation and some parts of the neocortex (stages III and IV), followed by most of the neocortex (stages V and VI). Individuals at stages I and II are asymptomatic, some individuals at stages III and IV show signs of memory impairment, and those at stages V and VI suffer AD (Schöll et al., 2016).
Amyloid-beta Peptide and Tau Protein Interplay:More Than Casual Partners
Currently it is becoming widely accepted that Aβ is the “trigger”and tau the “bullet” driving AD (Bloom, 2014). In addition, and although tau load better relates to cognitive decline than Aβ load, as mentioned, it has recently been reported that the presence of both Aβ and tau is necessary for memory decline in the preclinical stages of AD (Sperling et al., 2019). However, the mechanisms behind Aβ and tau interplay in AD remain elusive.
Recent data supports the hypothesis that there is a bidirectional interplay between Aβ and tau. Few years ago, it was demonstrated that addition of Aβ to human cells induces tau aggregation in the form of paired helical filaments-like filaments (Ferrari et al., 2003).Interestingly, cellular models have shown that over-expression ofAPPandPSEN1increases not only Aβ, but also tau aggregation (Choi et al., 2014). A relation between Aβ and tau was also found in animal models, as in APPPS1 transgenic mice (APP KM670/671NL (Swedish),PSEN1 L166P) overexpressing Aβ also show an increase in CSFtau levels. Furthermore, tau is able to facilitate Aβ aggregation, as tau deletion in APPPS1 transgenic mice ameliorated Aβ deposition(Peters et al., 2019); whereas the addition of human tau increased plaque size, supporting the hypothesis that there is a bidirectional interplay between Aβ and tau. However, the APPPS1 mouse model of AD does not develop NFTs, neither do other APP and/or PS1 models(Esquerda-Canals et al., 2017b). The only mouse model developing NFTs is the triple transgenic mouse model (3xTg-AD;PS1M146V,APPSwe,tauP301L) and this is so because one of the three transgenes is a mutated MAPT (tauP301L), found in familial FTD (Oddo et al., 2003).In addition, this mouse model is the only one where an evident neuron loss has been quantified even at the early stages of the disease. Interestingly, cellular density showed a strong correlation with intracellular Aβ burden in glutamatergic neuronal populations(Esquerda-Canals et al., 2017a). At the most advanced stage of the disease, it has been demonstrated that Aβ-immunotherapy not only decreases Aβ-burden but also affects tau load in this mouse model(Roda et al., 2020). Similarly, active DNA Aβ42immunization in 3xTg-AD mice reduced not only Aβ deposition but also tau pathology(Rosenberg et al., 2018).
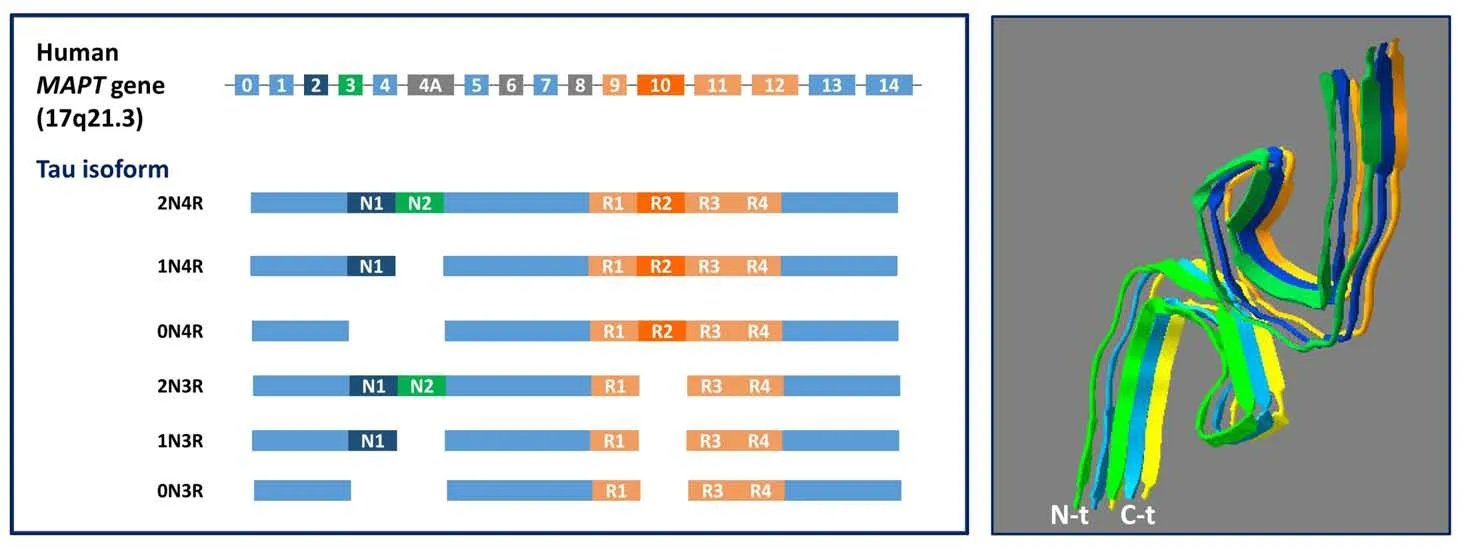
Figure 4 |Three-dimensional structure of tau NFTs and tau isoforms scheme.(A) Six different isoforms of tau are expressed in the adult human central nervous system via alternative splicing of the MAPT gene. Regulated expression of exons 2 and 3 gives rise to tau isoforms with 0, 1, or 2 N-terminal inserts whereas exclusion or inclusion of exon 10 leads to expression of tau isoforms with three (3R) or four (4R) microtubule-binding repeats. (B) Paired helical filaments from AD brain. The structure shows the fuzzy core formed by aggregated repeats (residues G304-E380) (PDB 6HRE)(Falcon et al., 2018). Each color represents a monomer.Images were generated with the Swiss-PdbViewer software (Guex and Peitsch, 1997).AD: Alzheimer’s disease; MAPT: microtubule associated protein tau; PDB: protein data bank.
The challenging modeling of AD in rats has been achieved with the line TgF344-AD expressingAPPSweandPS1∆E9genes (Cohen et al., 2013). This rat model shows Aβ oligomers, Aβ plaques, tau pathology, behavioral impairment and neuron loss. Abundant NFTs were detected in close proximity to β-amyloid plaques in aged transgenic rats that were reminiscent of NFTs found in AD patients brains. Taken into account that rats are 4–5 million years closer to humans than mice in evolution and that they express all six tau isoforms found in humans, this rat model supports the validity of the ACH in humans. Several studies show that Aβ and tau co-localize in neurons, as well as in astrocytes (Pickett et al., 2019). Aβ peptide is mainly accumulated in the synapses near to senile plaques, whereas tau synaptic localization is no dependent on senile plaques vicinity.Moreover, only 0.02% of the synapses were positive for Aβ and tau,indicating that direct Aβ-tau interaction might not be the major link between both pathologies. Interestingly, different crossings of MAPTnull + APP/PS1 + rTg21221 AD mouse lines (APP/PS1 + Tau),homozygous for deletion of mouse tau and heterozygous for the human wild-type tau inducible transgene, have shown that Aβ and tau cooperatively work to impair transcription of genes involved in synaptic function. In addition, accumulation of tau was observed in synapses of this mouse model and in human postmortem tissues.Downregulation of tau in these mice after behavioral deficits emerged, restored behavioral deficits, reduced synaptic tau levels,and partially reversed transcriptional perturbations (Pickett et al.,2019). Therefore, this is another evidence of tau being the effector molecule in AD.
Another proposed link between Aβ and tau alterations is the C-terminus of hsp70-interacting protein, as it has been reported that Aβ accumulation decreases C-terminus of hsp70-interacting protein expression and increases tau levels. In addition, and Aβinduced effects on tau are rescued by restoring C-terminus of hsp70-interacting protein levels (Oddo et al., 2008; Lyon and Milligan,2019).
A common hallmark among neurodegenerative diseases is neuroinflammation. The innate immune system could be another link between amyloid and tau pathologies. Interestingly, both Aβ and tau induce a neuroinflammatory response (Yoshiyama et al., 2007;Alawieyah Syed Mortadza et al., 2018) that leads to neuronal death,plasmatic leakage, and brain atrophy. On the one hand, microglia and complement factors seem to be involved in the early synaptic loss induced by oligomeric species of Aβ (Hong et al., 2016). On the other hand, some studies suggest that microglia could play a pivotal role in tau spreading by making synapses more vulnerable to tau-induced synaptic impairment. Inhibition of exosome secretion by microglia suppressed tau propagation to the dentate gyrus in a mouse model,recovering non-pathological excitability in this brain region. In contrast, extracellular soluble, but not intracellular, Aβ has been related with increased exosome-dependent tau spreading in both animal models and human (Gomes et al., 2019). These observations support the important role microglia, and particularly microglial exosomes, play in prion-like spreading of tau throughout the brain(Asai et al., 2015). It is worth noting that the immune system is not only a mediator between Aβ or tau and neuronal death. Aβ oligomers could activate the immune system, prior to senile plaque formation, triggering tau hyperphosphorylation and, subsequently,the formation of NFTs. Aβ oligomers are able to activate the inflammasome (Nod-like receptor family pyrin domain containing 3,NLRP3) that, in turn, regulates kinases, as glycogen synthase kinase 3(GSK-3) and phosphatases, as protein phosphatase 2A, inducing tau abnormal phosphorylation (Ising et al., 2019) and evidencing a clear link among Aβ, the immune system, and tau pathology.
Despite the evidence that the bidirectional interplay between Aβ and tau better explains the ACH, it is also true that there are factors that could independently affect and trigger both Aβ and tau abnormalities (Small and Duff, 2008). This idea has been formulated as “The dual pathway hypothesis”, which states that there are common upstream triggers causing both Aβ and tau abnormalities(Small and Duff, 2008). Supporting this hypothesis, it has been shown that Aβ is necessary but not enough to induce tau pathology. Aβ plaque-bearing mouse models that do not overexpress tau need injection of misfolded AD-tau seeds to develop the three major types of AD-relevant tau pathologies: dystrophic neurites surrounding Aβ plaques (NP tau), neuropil threads and NFTs. These distinct tau pathologies show different temporal onsets and functional consequences on neural activity and behavior (He et al., 2018).Among other factors,APOE, specifically the apoE ε4 isoform, would have an important role in this new scenario. ApoE ε4 increases the activity of one of the major tau kinases, GSK-3 (Liang et al., 2019),in neurons. Interestingly, GSK-3 is also involved in other dementias,as in FTD (Schaffer et al., 2008), and its inhibition reduces tau phosphorylationin vitro(Singh et al., 1995; Wagner et al., 1996),as well as in animal models (Uno et al., 2009). Thus, apoE or GSK-3 independent-Aβ alterations could enhance tau phosphorylation and aggregation, supporting the dual pathway hypothesis. Furthermore,it has been proposed that GSK-3 could be a linker between Aβ and tau hyperphosphorylation (Medina and Avila, 2012). Regarding Aβ,it has been widely reported that apoE ε4 enhances Aβ production and aggregation as well as impairs its clearance (Roda et al., 2019).Recently, an interaction effect between apoE ε4 and Aβ, rather than the sum of their independent effects, has been related to increased tau load in humans (Therriault et al., 2020). However, not only apoE ε4 is a common trigger of Aβ and tau aggregation. Several metabolic diseases have been suggested to be related with neurodegeneration.In fact, diet-induced insulin resistance in animal models stimulates Aβ and tau aggregation, as well as neurodegeneration (Bharadwaj et al., 2017). Which are the exact mechanisms behind such a stimulation remains to be elucidated, but the occurrence of amylin (or islet amyloid polypeptide) deposits in senile plaques suggests that a link between this pancreatic amyloid and AD could exist. Moreover,immune system dysregulation and oxidative stress, both present in type-2 diabetes, are involved in AD progression.
Thus, a large amount of evidence supports the assumption that Aβ peptide and tau protein are not casual partners. There is a link between both pathologies that cooperate to produce synaptic dysfunction, neuronal loss, and the eventual cognitive decline characterizing AD.
Combined Therapies as a New Hope: Targeting Amyloid-beta and Tau
Even though there is a bidirectional interaction and some factors could independently affect both molecules, Aβ oligomerization seems to precede tau hyperphosphorylation. The fact that Aβimmunotherapy still raises some concerns is in agreement with the new idea of Aβ acting just as a switch-on. Once Aβ triggers the cascade of events that leads to neuronal death and dementia,it is not that important whether its levels can be reduced or not because the effector molecule driving to synaptic impairment and neuronal death is tau. This would explain why previous clinical trials for Aβ-immunotherapy enrolling mild to moderate AD patients, did not reach the end-points. Nowadays, active clinical trials for Aβimmunotherapy enroll patients in early, even prodromal stages of the disease (see below).
In the last years, it has been reported that Aβ-immunotherapy reduces not only Aβ but also tau levels in animal models (Roda et al.,2020) and clinical trials (Blennow et al., 2012; Sevigny et al., 2016).Likewise, it has been shown that tau-immunotherapy also reduces Aβ levels (Dai et al., 2017). Since the interplay between Aβ and tau becomes evident, several researchers claim that a combined therapy would be a better strategy to treat AD at early stages of the disease(Busche and Hyman, 2020; Tolar et al., 2020).
Currently, there are three principal therapeutic approaches focused on Aβ: avoiding Aβ aggregation, reducing Aβ production, and enhancing Aβ clearance (Table 1). Only one drug intended for avoiding Aβ aggregation, Tramiprosate (reformulated as ALZ-801),has reached phase III but beneficial effect was only observed inAPOE ε4/4carriers (Abushakra et al., 2017; NIH, 2021). Other Aβ aggregation inhibitors, such as ELND005, did not reach primary outcomes in phase II (Rafii et al., 2017; NIH, 2021). Most secretases inhibitors/activators have reached phase III but safety concerns occurred in most of them (Vellas et al., 2012; Penninkilampi et al.,2016; Rombouts et al., 2021). The first active immunotherapy clinical trial for AD tested the AN1792 vaccine, composed of fibrillar Aβ (1–42) and QS21 adjuvant. Although phase II was discontinued because 6% of immunized patients suffered meningoencephalitis, clearance of plaques and reduction in cognitive decline were demonstrated (Vellas et al., 2009). Next generation of Aβ active vaccines have consisted of N-terminal Aβ sequences (i.e., 1–6, 1–14) encapsulated in different delivery systems (Hull et al., 2017; Lopez Lopez et al., 2019), The rationale behind the use of the N-terminal sequence is found in the 3D-structures of trimers (1–40) and dimers (1–42), both solvent exposing the N-termini, as mentioned. This rationale was also used for obtaining some of the monoclonal antibodies (mAbs) for passive Aβ-immunotherapy (La Porte et al., 2012; Salloway et al., 2014;Logovinsky et al., 2016; The Lancet Neurology, 2017; Manolopouloset al., 2019; Servick, 2020; Yoshida et al., 2020). Bapineuzumab, an humanized mAb, was the first reaching phase III clinical trials, but amyloid related imaging abnormalities were observed, mainly inAPOE ε4carriers, and no effect on cognitive decline was achieved(Salloway et al., 2014). Apart from the accelerated approval for aducanumab mentioned above, gantenerumab and BAN2401 have
potential for near term approval (Tolar et al., 2020). Aducanumab and gantenerumab are fully human mAbs that target epitopes composed of the N-terminus and a central region of the Aβ peptide,whereas BAN2401 is a humanized mAb directed to an epitope on the fibrils made by the E22G “arctic” mutation.
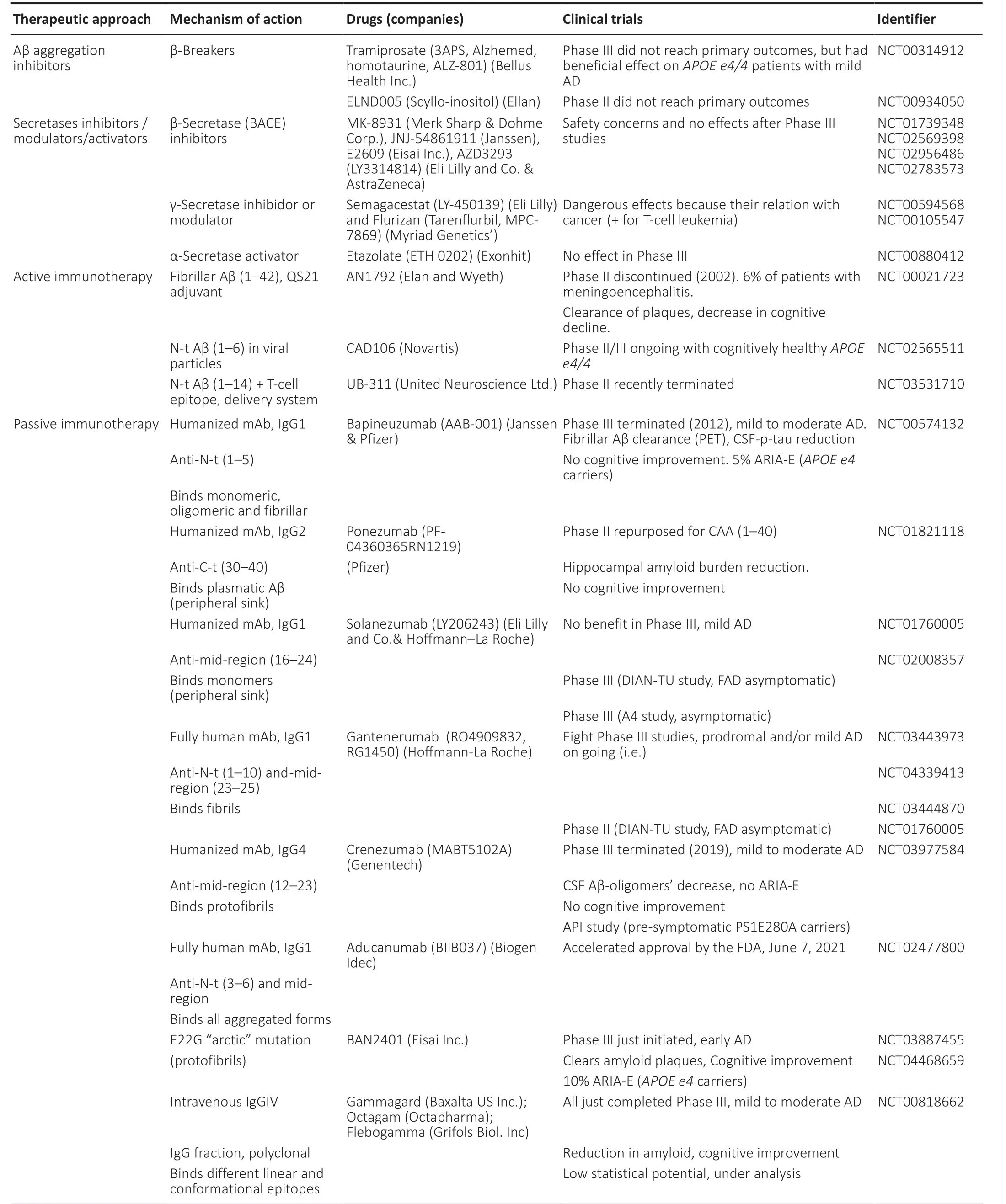
Table 1 |Aβ therapeutics in clinical trials
Similarly, treatments to reduce tau aggregation or decrease tau levels are under development (Table 2), but not as advanced as for Aβ. A new therapeutic approach for treating AD or other taupathologies is the use of antisense RNAs interfering the expression ofMAPTmRNA (Evers et al., 2015). Morpholinos are synthetic RNA analogs that are stable and specifically bind to the designed target mRNA. In neuroblastoma cell cultures, reductions in tau levels up to 80% were achieved by exon skipping inducing an out-of-frame deletion in tau mRNA (Sud et al., 2014). As mentioned before, six tau isoforms are found in the human brain (Figure 4A) and the ration between R3 (not including exon 10) and R4 isoforms (including exon 10) is impaired in several diseases such as PSP, Corticobasal Degeneration, Argyrophylic grain disease, Pick’s disease, and FTD-P, among others. Although this is not the case in AD, it is worth mentioning the capability of antisense oligonucleotides for controlling the skipping of exon 10 and therefore their therapeutic potential (Peacey et al., 2012). In this sense, a phase I clinical trials with the oligonucleotide NIO752(Novartis) aimed to treat PSP is being initiated (NCT04539041) (NIH,2021). In the specific case of the treatment of AD, IONIS MAPTRx oligonucleotide is being tested in phase I/II for decreasing tau mRNA(NIH, 2021). Another approach is the inhibition of tau aggregation that is being tested in a phase III clinical trial with methylene blue(Rember), already used for malaria. Concerning the enhancing of tau clearance, it must be taken into account that tau oligomers can exist in a variety of states including hyperphosphorylated forms, which can be both soluble and insoluble. In addition, the fact that they are six isoforms complicates the election of the best target. It remains to be determined (a) which of these oligomeric species of tau are causally involved in neurodegeneration, and (b) which trigger the beginning of the formation paired helical filaments filaments. Meanwhile,phase II trial for active immunotherapy with a fragment of tau(294–305) (AADvac1 (Axon Neuroscience SE) and several phase II trials for passive immunotherapy with gosuranemab, tilavonemab,semorinemab zagotenemab are ongoing (Alzforum, 2020). All these humanized mAbs have been obtained on an IgG4 scaffold and recognize the N-terminus of tau and hence extracellular tau, with the exception of zagotenemab that also recognizes a mid-region characterizing aggregated tau.In conclusion, the evidence of a bidirectional interplay between Aβ and tau better explains the ACH, which is currently complemented by the dual pathway hypothesis. Combined anti-Aβ and antitau therapies will be tested in the near future because Aβimmunotherapy is quite advanced, with a recent accelerated approval for Aduhelm, and tau-immunotherapy is on the path. In addition, the determination of the three-dimensional structures of the fibrils made by Aβ1–40, Aβ1–42, and tau may be helpful in the design of new therapeutic molecules. Therefore, it is quite likely we are on the path to find a cure for this devastating disease.
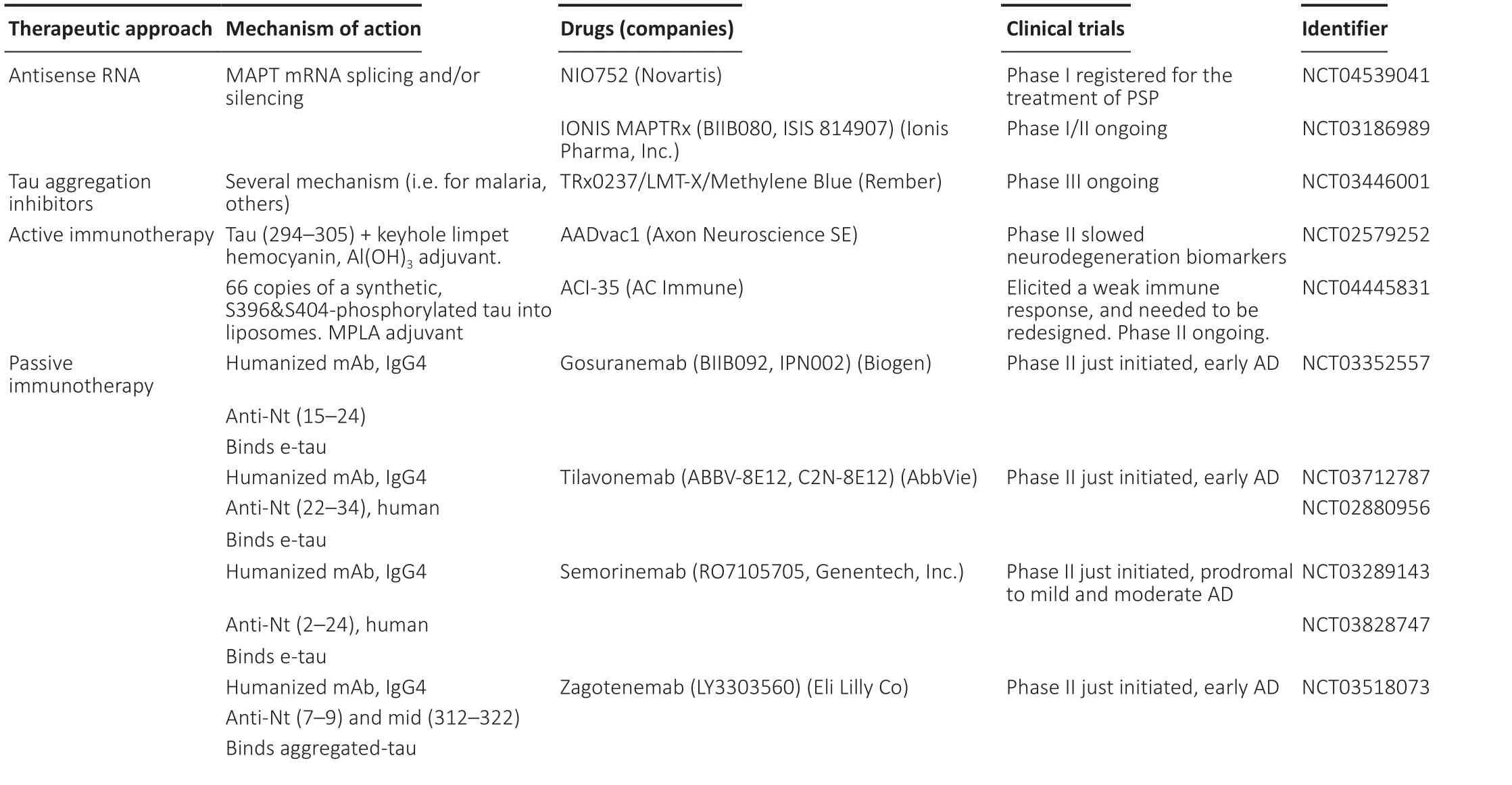
Table 2 |Tau therapeutics in clinical trials
Author contributions:ARR designed the manuscript; ARR, GSM, LMG and SV collaborated in data search and manuscript writing; LT participated in manuscript revision; SV performed final manuscript revision. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long asappropriate credit is given and the new creations are licensed under the identical terms.
©Article author(s) (unless otherwise stated in the text of the article) 2022.All rights reserved. No commercial use is permitted unless otherwise expressly granted.
杂志排行
中国神经再生研究(英文版)的其它文章
- Ocular therapies for neuronal ceroid lipofuscinoses: more than meets the eye
- Designing nanocarriers to overcome the limitations in conventional drug administration for Parkinson’s disease
- The second brain in Parkinson’s disease: fact or fantasy?
- Elevated intraspinal pressure in traumatic spinal cord injury is a promising therapeutic target
- Construction and imaging of a neurovascular unit model
- Phytochemicals as inhibitors of tumor necrosis factor alpha and neuroinflammatory responses in neurodegenerative diseases