单病毒示踪技术在病毒侵染机制及水生动物病毒研究中的应用进展
2022-01-10秦启伟
秦启伟
(华南农业大学 海洋学院,海洋生物资源保护与利用粤港澳高校联合实验室,广东省水产免疫与健康养殖工程技术研究中心,广州 510642)
病毒是一类结构简单的微小生物,一般是在蛋白质外壳内包裹一种脱氧核糖核酸(DNA)或核糖核酸(RNA)作为遗传物质。病毒没有细胞结构,无法独立生长及复制,只有借助宿主细胞才能完成自身的复制和繁衍,是严格的细胞内寄生物[1]。病毒尽管结构和组成极其简单,却是对人类及其他动物生存威胁最大的病原之一。2019年12月爆发的SARS-CoV-2导致的新型冠状病毒肺炎(Corona virus disease 2019, COVID-19),仅用2个月就席卷全球,截至2021年10月6日,全球累计感染约2.37亿人,死亡人数超483万。人类免疫缺陷病毒(Human immunodeficiency virus,HIV)、乙肝病毒(Hepatitis B virus,HBV)、非洲猪瘟病毒(African swine fever virus,ASFV)和禽流感病毒(Avian influenza virus,AIV)等病毒也严重威胁人类健康、动物安全和社会经济发展等。鱼类等水生动物栖息在水中,环境复杂多变,深受病毒感染的威胁。目前,已从养殖或野外鱼类当中分离到多种病毒性病原,如神经坏死症病毒(Nervous necrosis virus,NNV)、石斑鱼虹彩病毒(Singapore grouper iridovirus,SGIV)、传染性脾肾坏死病毒(Infectious spleen and kidney necrosis virus, ISKNV)和草鱼呼肠孤病毒(Grass carp reovirus, GCRV)等[2]。这些病毒已成为严重威胁水产健康养殖的最重要病原之一,病毒性疾病一旦暴发,无法有效治理,易造成重大经济损失,危害严重。因此,对病毒侵染宿主过程的诠释是认识、预防及控制病毒性疾病暴发的重要途径。
病毒侵染细胞是一个多步骤且与多种细胞结构相作用的极复杂过程,主要包括进入、运输、脱壳、复制、子代病毒组装和释放等一系列步骤[3]。如图1所示,病毒侵染起始于病毒与细胞膜的黏附接触,一般通过多糖-蛋白复合物的低亲和静电相互作用,继而病毒与受体特异性结合,并通过膜融合或内吞进入细胞,进入方式和机制复杂多变。病毒进入细胞之后,常常被分选至内体囊泡中,并借助细胞骨架运输到细胞核或近核区等特定部位,释放病毒核酸,启动病毒基因复制和蛋白表达。这些新合成的病毒蛋白和病毒核酸逐步成熟并被运输到病毒装配位点,组装完成的子代病毒通过出芽、胞吐或裂解细胞而释放到细胞外,又可感染新的宿主细胞[2]。
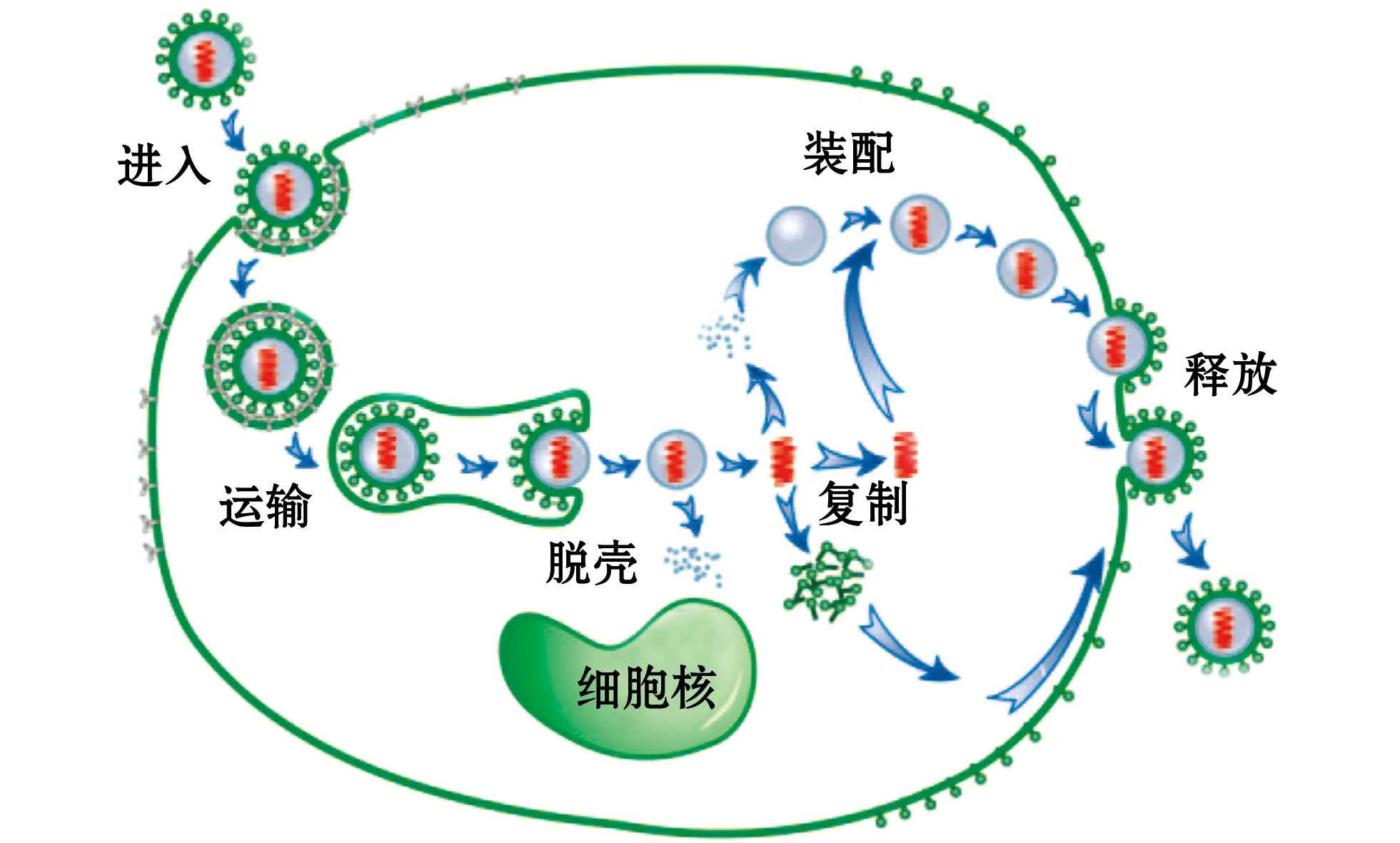
图1 动物病毒生命周期(修改自Gao等)[4]Fig.1 Animal virus life cycle(modified from Gao et al)[4]
病毒侵染细胞,不仅病毒与细胞之间的相互作用是动态的,而且细胞自身也处于不断动态变化中[5]。通过传统生物化学、分子生物学和透射电子显微镜等技术,可以分析病毒在某个特定时间、空间、平均化的感染情况,但是难以获得病毒侵染的动态过程和详细机制。随着生物物理学、化学和材料科学的快速发展及在生物学的渗透,研究非均匀体系实时动态变化的单病毒示踪(Single-virus tracking,SVT)技术的建立,使得研究人员能够追踪单个或多个病毒粒子的侵染过程,可视化、实时检测病毒-细胞相互作用的动态过程,更加全面精细地阐述病毒侵染的时空动态过程及机制。
虽然单病毒示踪技术目前主要应用在高等哺乳动物病毒研究中,但是已开始逐步应用于鱼类病毒感染机制研究中。本文中将系统介绍单病毒示踪技术的方法体系,总结该技术在病毒特别是鱼类病毒感染机制研究中的成果,并对该技术应用于病毒学研究的发展趋势进行展望。
1 单病毒示踪技术体系
从20世纪80年代开始,基于光学显微镜的单病毒示踪技术使人类能够实时亲眼“看到”病毒如何感染宿主细胞,将人类的视野聚焦到单粒子水平病毒的可视化研究。1980年,Helenius等[6]首次在固定的动物细胞中检测到单个荧光标记的西门利克森林病毒(Semliki forest virus, SFV)。1981年,Barak等[7]实时追踪活细胞中的单个低密度脂蛋白(Low-density lipoprotein, LDL)-受体复合物,这是首次报道在活细胞中进行的单粒子示踪(Single-particle tracking, SPT)试验。1985年,发展出了更适合二维SPT的成像技术和图像分析算法,为单病毒示踪技术奠定了基础[8-13]。1988年, Bachi等对单个呼吸道合胞体病毒(Respiratory syncytial virus, RSV)的出芽和融合过程进行了实时观察[4]。1990年,Lowy等观察了单个流感病毒-细胞融合[5]。同年,Georgi等利用荧光显微镜在活细胞中检测到单个呼肠孤病毒(Reovirus serotype 1)并追踪了其侵染细胞的早期阶段[6]。此后,多色荧光标记病毒粒子的示踪、三维空间单病毒示踪技术及动物体内的病毒示踪等相继被报道,单病毒示踪技术成为研究病毒侵染机制的有力工具[7-20]。
单病毒示踪是一种使用荧光显微镜监测活细胞中的单个病毒粒子或病毒组分的实时成像技术(图2),是SPT技术在病毒学领域的具体应用。借助单病毒示踪技术,研究人员可以观察单个病毒粒子或病毒组分与活细胞相互作用的动态过程,实时获取病毒侵染过程中重要或关键分子事件[21-22]。例如,病毒如何启动进入,在病毒进入宿主细胞的过程中相关宿主蛋白在空间和时间上发生怎样的变化,病毒基因组释放在细胞内何时何地进行等。这些信息的获得能够最本质和最真实地揭示病毒侵染和致病等生命活动,对病毒学研究具有重大意义。

图2 单病毒示踪荧光显微成像系统[5]Fig.2 Fluorescence microscopes for single-virus imaging[5]
1.1 荧光标记单粒子
要成功示踪单个病毒粒子,首先必须对目标粒子和感兴趣的细胞结构进行足够数量且不影响生物活性及细胞功能的荧光标记。能够进行标记的理想荧光探针应具有的特点包括耐光和不易发生光漂白,高吸光系数和高量子产率,易被激发,荧光强度波动小,体积小,以及对标记的病毒或宿主影响小等。在活细胞中,较长激发和发射波长的荧光探针可避免激发光的光毒性,且易与细胞自发荧光信号区分。目前,病毒标记常用的荧光标记物主要包括有机荧光染料、荧光蛋白和量子点等。
1.1.1 有机荧光染料 有机荧光染料是含有共轭双键、苯环等结构的小分子化合物,具有尺寸小、易合成、种类多(基本覆盖了从紫外到近红外的光谱范围)等优势,在荧光成像领域应用广泛。常见的荧光染料包括氰色素类(如Cy2、Cy3、Cy5)、Alexa Fluor染料家族(如Alexa Fluor488、Alexa Fluor598、Alexa Fluor647)、亲脂性染料(如DiO、DiI、DiD)等,以及一些具有特殊性质的有机荧光染料,如pH敏感荧光染料(CypHer5)和靶向核酸染料(SYTO和Hoechst系列)等。Seisenberger等[23]通过共价化学反应用Cy5染料标记腺病毒的衣壳,详细阐述了该病毒在活细胞内的侵染过程。Van der Schaar等[24]和Lakadamyali等[25]用亲脂性染料DiD标记病毒包膜,分别研究了流感病毒、登革热病毒(Dengue virus,DenV)的侵染路径。
1.1.2 荧光蛋白 绿色荧光蛋白(Green fluorescent protein,GFP)是由下村修在20世纪60年代早期从维多利亚多管发光水母中发现并纯化[26]。经过数十年的发展,荧光蛋白的光谱已经覆盖从紫外到近红外光谱,广泛应用于生物成像领域[27-28]。Rietdorf等[29]利用GFP标记的牛痘病毒(Vaccinia virus, VV)观察了其从病毒组装的近核位点到细胞膜的运动。除了常见的GFP、YFP和RFP等荧光蛋白,研究人员还开发出许多特殊性质的荧光蛋白,如pH敏感的荧光蛋白、光激活荧光蛋白和双分子互补荧光蛋白等。Hogue等[30]用pH敏感GFP标记伪狂犬病毒(Pseudorabies virus, PRV)并追踪了其运动和胞吐。
1.1.3 量子点 量子点(Quantum dots,QDs),又称为发光半导体纳米微晶体,最早是在20世纪80年代制备得到,其光谱可以随量子点的大小、形状和组成的变化而变化,目前已经覆盖蓝色到近红外的光谱范围。量子点具有独特的光学性质(单一波长的光可同时激发多颜色的量子点)、较大的斯托克斯位移、荧光强度高、抗光漂白能力强、荧光寿命较长等优势,比传统的有机染料发射荧光更强、光稳定性更好,在荧光成像方面展现出极大的发展潜力[31-32]。Liu等[33]利用QDs标记流感病毒,并在三维空间尺度追踪了与Rab5和Rab7相关的病毒感染过程。Lü等[34]用QDs标记PRV并进一步分析了病毒进入细胞的主要途径。
1.2 荧光显微镜成像系统
标记好的病毒粒子需在具备检测器灵敏度高、成像速度快、激发体积小等性质的荧光显微镜下观察,目前,落射荧光显微镜(Epifluorescence microscopy)、共聚焦显微镜(Confocal microscopy)和全内反射荧光显微镜(Total internal reflection fluorescence microscopy)是最常用的3种荧光显微镜装置[5]。
1.2.1 落射荧光显微镜 落射荧光显微镜结构简单,利用平行光束照射大范围样品区域,并用高数值孔径物镜收集来自样品的发射光,具有成像景深大、信号损失小、快速宽场检测等优点,可用于长距离追踪病毒运动[5,35]。由于成像景深大,且细胞本身有自发荧光会产生背景噪音,落射荧光显微镜成像的信噪比低,不适合对荧光信号较弱的病毒粒子进行追踪。
1.2.2 共聚焦显微镜 共聚焦显微镜是单病毒示踪中常用的一种成像装置。共聚焦显微镜在普通的显微镜基础上,以激光作为光源,在光路中加入了相对于物镜焦平面共轭的照明针孔与探测针孔,使样品中只有处于焦平面的部分能够被激发光照射,且只有焦平面处发射的荧光被光检测器接收,有效地降低了成像的背景噪音,同时实现Z轴方向不同焦平面的连续成像,完成对样品的三维成像。但是,这种成像技术也会导致信号损失。常用的共聚焦显微镜主要包括点扫描式和转盘式两种。点扫描式共聚焦显微镜通过激光扫描头逐点逐行地扫描样品,成像速度较慢。转盘式共聚焦显微镜在点扫描式共聚焦显微镜的基础上发展而来,其光路中使用多个微透镜-针孔聚焦模组构成的阵列替代传统的单个透镜-针孔聚焦模组,并置于高速旋转的转盘上,实现多通道检测成像,同时,利用高灵敏的电荷耦合器件(Charge coupled device,CCD)对荧光信号同时采集,有效地提高了成像速度[36-37]。
1.2.3 全内反射荧光显微镜 全内反射荧光显微镜属于宽场成像技术,以入射光(角度足够大)照射在两个折射率不同的介质的临界面(如细胞和玻璃基底的界面)发生全内反射时产生的消逝波作为激发光,激发靠近临界面区域约200 nm以内的荧光信号,从而有效避免样品其他区域的荧光干扰,提高信噪比和成像质量。然而,该成像技术也因此受限于研究接近细胞表面的生命活动[38]。
此外,近年来快速发展的突破光学衍射极限的超分辨荧光显微镜,如受激发射损耗显微镜(Stimulated emission depletion microscopy,STED)、结构照明显微镜(Structured illumination microscopy,SIM)、光激活定位显微镜(Photoactivation localization microscopy,PALM)和随机光学重构显微镜(Stochastic optical reconstruction microscopy,STORM),使人们能够在更微小的尺度追踪病毒的生命过程[22]。
1.3 荧光图像数据分析
活细胞成像获得的系列图像,每一组可能由成百上千帧图像组成,而每一帧图像包含上百万字节的数据,需要通过一系列的计算机算法在大量的图像信息中进行图片信号处理、数据统计等,从而获得病毒粒子的运动轨迹、运动速率、运动方式等[5]。这一系列的图像处理分析方法,主要包括降低图像背景噪音、病毒颗粒的精确定位、轨迹重建和轨迹分析等(图3)。

图3 单病毒示踪图像处理过程(修改自Brandenburg等)[5]Fig.3 Procedure of image analysis of single-virus tracking technology(modified from Brandenburg et al)[5]
1.3.1 粒子定位 在试验条件下,单个病毒粒子的荧光信号与背景信号和噪音相比较弱,使得背景信号和噪音容易被作为假性峰值检测到,因此,获得的原始图像首先要经过空间滤波器如二维高斯卷积算法来降噪,然后再对图像中的病毒粒子进行高精度定位[39-40]。目前,已经发展出数种高精度的粒子定位算法如反卷积技术、二维高斯拟合和质心法等以精确地确定粒子的位置,这些算法大多基于点扩散函数实现光斑定位[41-42]。二维高斯拟合法是最常用的一种算法,对光斑的定位精度高,可以精确到几纳米,但是需要反复迭代,计算速度慢。质心法计算简单且速度快,但是容易受背景噪音干扰,定位精度较低。
1.3.2 粒子轨迹重建 轨迹重建就是将同一个颗粒在不同帧图像中的坐标按时间顺序连接在一起。这一步最大的困难是需要保证所连接的坐标确实来自同一个颗粒,因为同一张图像中多个粒子可以被定位,距离较近的粒子会干扰判断,并且这些粒子在活细胞内的运动往往非常复杂,包括粒子碰撞、融合、分裂、跳跃和闪烁等,这些问题导致粒子运动轨迹重建的步骤非常复杂。目前,已有不同的自动化算法可实现轨迹重建[43-44]。这些算法通常利用粒子光斑大小、距离、荧光强度等参数来判断不同帧图像的病毒粒子是否为同一粒子。Jaqaman等[44]基于代价矩阵建立了多假设算法来解决复杂的光斑合并、拆分与闪烁等问题。
1.3.3 粒子轨迹分析 对重建的轨迹进行分析,可以获得病毒的运动速率、均方位移、扩散系数,以及与其他蛋白或细胞结构之间共定位时间、荧光强度的变化等,从而定量地获得一系列病毒运动相关的参数。粒子的瞬时运动速度是粒子运动相关的重要参数之一,基于重建的粒子轨迹,可以获得粒子在不同帧图像间的运动距离,结合拍摄图像的时间间隔,就可以获得粒子在不同时间点的瞬时运动速度。另外,根据粒子的均方位移(mean-square displacement, MSD,即粒子在一段时间内平方距离的平均值)与时间间隔的依赖关系可以对粒子运动模式进行分类,进一步获得粒子运动的扩散系数、拟合速度等参数。粒子运动模式主要包括扩散运动、定向运动、非正常扩散运动和受限运动[45-46]。
除了获得粒子运动速度、运动模式和扩散系数等外,病毒粒子在不同帧图像的荧光强度随时间的变化规律、聚集和解聚以及与其他荧光信号的定位情况等也可以被进一步分析。
2 单病毒示踪在病毒学研究中的应用
目前,国内外许多课题组,包括庄小威、庞代文、崔宗强和王宏达等许多学者带领的研究团队,推动了单病毒示踪技术的快速发展和应用,使人类“看到”病毒如何感染宿主细胞。
2.1 病毒黏附
一些病毒如鼠白血病病毒(Murine leukemia virus,MLV)、禽白血病病毒(Avian leukosis virus,ALV)、HIV、水疱性口炎病毒(Vesicular stomatitis virus,VSV)、小鼠多瘤病毒样颗粒(Murine polyoma virus-like particle,MPy VLP)、人乳头瘤病毒-16假病毒(Human papillomavirus-16 pseudovirus,HPV-16 PsV)、VV、1型单纯疱疹病毒(Herpes simplex virus type 1, HSV-1)和丙型肝炎病毒(Hepatitis C virus,HCV)等,到达细胞表面后,能够以依赖微丝的方式在细胞表面运动[47-52]。HPV-16 PsV在细胞表面具有4种典型运动模式,包括小区域(直径30~60 nm)内的受限运动、缓慢漂移的受限运动、突然终止的快速随机运动和长距离的线性定向运动。HPV-16 PsV的定向运动大多是在沿着细胞表面富含微丝的突起如丝状伪足或收缩纤维上进行的,且运动速度也与微丝收缩的速度相似[5]。甲型流感病毒(Influenza A virus)到达细胞表面也显示了4种不同的运动模式,包括受限、定向的弹射(速度高达1~2 μm/s)、漂移和混合模式等,并且病毒的不同运动模式受细胞表面黏附因子浓度的影响[53]。
通过常规的生物学分析法去了解病毒结合如何诱导受体聚集或者信号转导的详细机制是非常困难的,因为病毒和受体之间的相互作用常常发生在一瞬间。实时追踪犬细小病毒(Canine parvovirus,CPV)依赖网格蛋白的内吞过程,研究人员发现,大部分CPV衣壳蛋白结合的转铁蛋白受体(Transferrin receptors,TfR)小于5个,大约25%结合有TfR的病毒侧向扩散到达进入位点,76%的CPV由于病毒衣壳和细胞受体之间的低亲和力结合而在内吞之前脱离细胞膜[54]。
2.2 病毒进入
通过单病毒示踪技术,能观察病毒进入活细胞的精细过程。流感病毒(Influenza virus)和VSV诱发网格蛋白从头组装[39,55-56],而CPV和DenV则进入细胞膜上已经装配好的网格蛋白有被小窝(Clathrin-coated pit, CCPs)[54, 57]。另外,流感病毒能够同时利用两种途径进入细胞:大部分病毒粒子促进CCPs的从头合成,从而通过网格蛋白介导的内吞进入细胞;而其余病毒粒子通过网格蛋白和小窝蛋白非依赖型的内吞进入细胞。通过这两种方式进入的流感病毒粒子表现出相似的运输过程,并且具有相似的基因释放效率[39]。
实时追踪SV40病毒粒子与GFP荧光标记的细胞膜微囊,结果表明,大多数SV40与细胞膜微囊共定位,激活酪氨酸激酶并重聚微丝形成actin tail位于包含病毒粒子的细胞膜微囊,经过动力蛋白依赖的颈部收缩后,包含SV40的细胞膜微囊离开细胞膜,沿着微管向内质网运动[58-61]。
2.3 病毒的胞内运输
病毒进入细胞之后,常常借助细胞骨架进行胞内运输。大部分病毒粒子依赖微管运动,HSV、流感病毒、SV40、HIV、SFV和手足口病毒(Foot and mouth disease virus,FMDV)等病毒粒子能以数微米每秒的速度沿微管运输[25,59, 62-66]。但也有少量的病毒只依赖微丝运动,并且具有不可思议的高速度。脊髓灰质炎病毒(Poliovirus,PV)只依赖微丝进行快速的不定向运动,运动速度高达5 μm/s[67]。乙肝病毒表面抗原(Hepatitis B surface antigen,HbsAg)也被发现只依赖微丝进行快速的胞内运动[68]。
很多病毒粒子的胞内转运过程同时依赖微管和微丝。流感病毒在细胞内的运动可被分为3个阶段,首先流感病毒粒子在细胞膜内边缘区域依赖微丝的运动,随后由动力蛋白介导的沿着微管向近细胞核区域的快速运动,最后在近核区沿着微管进行双向的往复运动[25]。同样地,在三维空间尺度实时追踪HIV病毒粒子的运动发现,HIV在胞内运动会依赖微管和微丝[66]。
2.4 病毒脱壳
病毒进入细胞后,要将自身基因组从衣壳的包裹中释放到细胞质特定位点或细胞核中,才能启动后续复制。用荧光染料Syto 82和CypHer5分别标记PV的RNA和衣壳,在荧光显微镜下实时观察病毒的基因组释放,发现PV的RNA释放只发生在病毒粒子进入细胞之后靠近细胞膜的囊泡中[21]。这个研究解决了长久以来有关PV的RNA释放位点的争论。Qin等[69]用量子点标记流感病毒的vRNP,追踪了单个病毒的脱壳过程,发现病毒8个vRNP在脱壳过程中以单独而非组团的方式从晚期内体释放到细胞质,每个vRNA均以典型的三阶段运动模式进入细胞核,而后又以两种不同的扩散模式运动到其复制位点。
大部分DNA病毒和一些RNA病毒在其基因组释放进入细胞质后必须经历核运输来启动病毒后续感染事件。腺病毒(Adenovirus)通过kinesin-1介导与NPC相作用,完全破坏衣壳释放DNA入核[70],而且只有6%~48%的病毒DNA能进入细胞核,即使增加感染细胞的病毒数量,进入细胞核的病毒DNA也不能随之增加,运输病毒DNA的位点可能是有限的[71]。含有FLAsH标记的整合酶的HIV在经过微管依赖的运动、微丝依赖的运动及核膜上的受限运动后,在细胞核内还将进行扩散运动[66]。研究者普遍认为,完整的HIV-1不能入核且脱壳发生在细胞核膜附近,然而,Burdick等[72]追踪GFP标记病毒衣壳蛋白的HIV-1脱壳过程时发现,完整(或近乎完整)的病毒会在脱壳前进入细胞核并完成逆转录,在基因组整合之前1.5 h内且靠近(<1.5 μm)基因整合位点处开始脱壳,丰富了人们对于HIV-1脱壳机制的认识。
2.5 子代病毒粒子的组装和释放
病毒复制的子代基因组如何运动是存在已久的问题。流感病毒在细胞核中完成基因组复制后,其vRNPs从细胞核通过循环内体向细胞膜附近运输[73]。Chen等[74]通过单粒子示踪发现,大部分HIV-1的RNA在细胞质中以不依赖细胞骨架的非定向、随机漫步的方式运动,并且不受病毒Gag(Group-specific antigen)蛋白是否表达的影响。
MLV倾向于在细胞之间的接触区域装配子代病毒,在细胞接触区域发生的病毒装配比非接触区域约高10倍[75]。细胞内具有囊膜的VV病毒粒子从核周的装配区域通过微管向细胞膜运动[5]。这些病毒粒子与细胞膜融合,成为与细胞连接的细胞相关囊膜病毒(Cell-associated enveloped virus)[76]。VV的囊膜蛋白B5R激活Src家族激酶,进而磷酸化病毒蛋白A36R,接着使微丝聚合形成典型的肌动蛋白尾,从而促进病毒在细胞之间的扩散[77]。装配完成的ASFV子代病毒粒子同样沿着微管向细胞膜运动,随后借助微丝聚合从细胞中释放[78-79]。
此外,Liu等[80]基于Halo Tag定点标记病毒衣壳的重组PRV并结合多样的Halo Tag标签蛋白荧光配体,对亲代病毒进入、胞内运输,以及子代病毒衣壳组装、在细胞核和细胞质扩散运动进行研究,完成了对PRV完整的生命周期的示踪。
3 单病毒示踪技术在水生动物病毒研究中的应用
除了哺乳动物病毒,单病毒示踪技术也逐渐应用于水生动物病毒侵染机制研究。本课题组利用小分子荧光染料对纯化的SGIV粒子进行荧光标记,在共聚焦显微镜下实时追踪SGIV侵染宿主细胞的动态过程。SGIV能够沿着细胞表面的丝状伪足样突起到达细胞表面后进入细胞(图4(a)),或者随着细胞表面突起的收缩而到达细胞表面(图4(b));进入细胞的病毒粒子分别与早期内体、晚期内体和溶酶体共定位,并沿微丝和微管运动,瞬时速度可达0.9 μm/s,分别破坏微丝和微管后,病毒粒子运动受到显著影响;同时结合特异性抑制剂和共定位分析,本课题组发现,SGIV进入细胞的方式与其他虹彩病毒明显不同,SGIV进入细胞通过pH、dynamin依赖的网格蛋白介导的内吞,而不通过小窝蛋白介导的内吞;并且首次发现虹彩病毒进入宿主细胞的途径亦包括巨胞饮(图4(c))[81]。
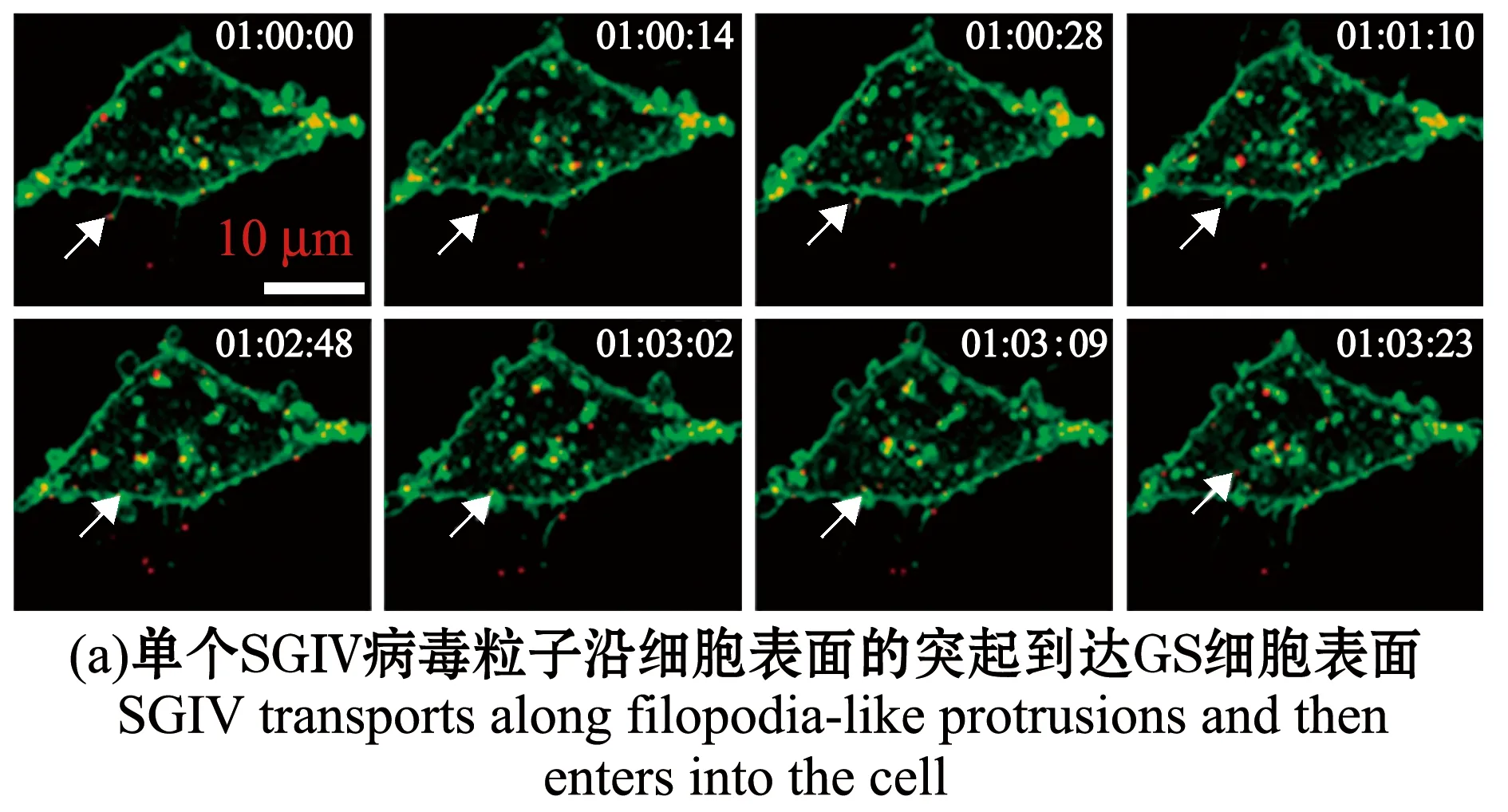
图4 SGIV侵染宿主细胞[81]Fig.4 SGIV infests host cells[81]
通过自主建立的基于原子力显微镜(Atom force microscopy, AFM)的微观生物力测量新方法——力示踪技术,首次研究SGIV侵染宿主细胞的动力学机制。病毒粒子进入细胞的位移峰值分别为(180.2±21.6)、(81.0±6.2)nm,分别对应于病毒粒子连接在探针尖端或侧面时进入细胞的情况;两种情况下,病毒粒子进入细胞的时间分别为(1.42±0.68)、(0.82±0.06)s,病毒粒子的最大瞬时速度约为200 nm/s。而应用恒定位置模式的力示踪技术能够测量单个SGIV病毒粒子进入宿主细胞的作用力,单个SGIV病毒粒子受到细胞的内吞作用力为(60.8±18.5)pN[82]。相关研究结果极大地丰富了对虹彩病毒进入方式的认识,也拓展了单病毒粒子示踪技术在水生动物大分子DNA病毒研究中的应用。
鱼类传染性造血组织坏死症病毒(Infectious hematopoietic necrosis virus,IHNV)、大菱鲆呼肠孤病毒(Scophthalmus maximus reovirus,SMReV)、草鱼呼肠孤病毒(GCRV)和对虾白斑综合征病毒(WSSV)等水生动物病毒分别被量子点或小分子荧光染料标记进行了单病毒示踪分析。量子点标记的IHNV 通过网格蛋白介导的内吞途径进入鱼类宿主细胞,在早期内吞小泡通过微丝运输进入胞浆,然后通过微管运输到达早期内体、晚期内体,最后到达溶酶体,IHNV沿微管运动可持续数秒,瞬时运动速度约0.59 μm/s[83]。量子点标记的SMReV结合到细胞膜上可以在数秒内通过新生的CCP进入细胞,且大部分病毒能够在感染30 min内以网格蛋白介导的内吞进入细胞,而后先借助微丝在近膜区运动而后依赖微管向细胞中心运动,运动速度约0.23 μm/s[84]。GCRV经过量子点标记后,同样被观察到沿微管运动[85]。DiO/DiD标记的WSSV在细胞中可能利用溶酶体进行运输[86]。
目前,应用单病毒示踪技术进行研究的水生动物病毒还比较少,尚属于起步阶段。
4 存在问题及展望
单病毒示踪技术已广泛应用于研究病毒感染宿主细胞的动态过程和机制,包括病毒进入、运输、基因组传递和子代病毒组装和释放等。作为一项多学科交叉发展起来的新技术,单病毒示踪技术的发展得益于化学、物理、计算机、细胞生物学和病毒学等多个领域研究人员的共同努力。随着这些学科的不断发展,单病毒示踪技术也处于持续快速发展中,然而单病毒示踪技术仍面临一些问题与挑战。
4.1 单病毒示踪技术研究存在问题与挑战
1)病毒标记物选择。大部分病毒粒子尺寸很小,且感染过程可能持续时间较长,就要求用于单病毒示踪的荧光标记物必须具备优异的性能,要求标记物荧光信号强、稳定性高、抗淬灭、无毒性、尺寸尽可能小、操作简便且不影响病毒感染活性。然而,目前常用的标记物仍存在不同的问题。有机荧光染料尺寸小、使用方便,可通过简便快速的操作与病毒囊膜、衣壳或核酸偶联,但荧光强度较弱、易淬灭且需要纯化病毒。并且对于尺寸很小(如粒子直径20~30 nm)的病毒,在不引起染料分子间自淬灭效应(self-quenching effects)和病毒感染活性的情况下,能够连接到病毒粒子的荧光染料数量是非常受限的。荧光蛋白可以通过常规的基因操作手段特异性地标记病毒成分,但操作繁琐、荧光强度较弱、尺寸较大且可能影响病毒活性。量子点荧光强度高、耐光漂白,但尺寸较大、存在非特异性吸附且可能影响病毒感染力。
2)制备多色标记病毒。病毒的完整生命周期包括进入、运输、脱壳、转录、复制、组装和释放,荧光标记的病毒如何在整个感染过程持续地以高分辨率被监测仍是一个亟待解决的问题。病毒的不同组分都可以被荧光标记,衣壳和囊膜有更多的标记物和标记方法可供选择,但是病毒内部组分如病毒核酸被荧光标记是比较困难的。只有衣壳或囊膜被标记的病毒粒子可以用于示踪病毒早期感染过程,但是不适于病毒感染过程的持续示踪,如观察病毒核酸复制、子代病毒粒子组装和释放等过程。因此,制备多色标记不同结构的病毒才能更好地对病毒感染过程进行持续地观察。如何高效地荧光标记病毒核酸、子代病毒,有效地区分亲代病毒和子代病毒,同时标记病毒的不同组分包括囊膜、衣壳和核酸,这些问题的解决仍有待进一步的探讨。
3)成像技术。病毒感染过程涉及大量的宿主蛋白和病毒蛋白之间的相互作用,有些作用过程是比较快速或稀少的,往往需要更高的时间分辨率和空间分辨率来高效捕捉相关过程。理想的成像技术具有成像快速、分辨率高、自动对焦、三维成像等性能。目前常用的荧光显微镜仍存在成像速度和分辨率较难同时兼顾,且仪器操作繁琐等问题。共聚焦显微镜操作相对简便,且适合活细胞成像,但受限于分辨率,无法清晰成像小于200 nm的结构。三维成像比二维成像能提供更多的信息,而三维成像也依赖于高时空分辨率的成像仪器。
4)活体成像。单病毒示踪技术目前主要应用于体外培养的细胞中示踪病毒,是一个简化的病毒感染模型。要充分了解病毒感染机制,需要进行动物体内试验。追踪病毒侵染动物活体的过程,有助于研究者探讨病毒如何突破机体的防御屏障,如何在不同组织不同细胞间传播等问题。然而,由于活体成像技术的局限,动物活体内单个病毒的实时追踪仍然是一个巨大的挑战。
5)图像信息分析。分析获得的图像数据,必须精确定位图像中的病毒粒子,并将同一个病毒粒子在每一帧图像中的位置连接,从而构建病毒粒子轨迹。然而,由于病毒在活细胞内的运动复杂,特别是当大量病毒感染细胞时,粒子之间的干扰增强,如何用合适的算法获得最真实的粒子轨迹,是一个不小的挑战。另外,病毒粒子从接触细胞表面开始到最终完成子代病毒粒子的组装和释放,要经历一系列复杂的步骤,并且与宿主细胞的多种蛋白、结构发生相互作用。在此过程中,各种各样的病毒运动轨迹可以被追踪到,而这些轨迹可能对应于不同的事件,可能是病毒无法突破细胞膜屏障进入细胞,可能是病毒进入细胞的不同方式,可能是病毒到达复制位点的“必经之路”,可能是病毒“误入歧途”而无法到达特定的复制位点。因此,病毒粒子的每一条运动轨迹都包含了大量的信息,如何快速、准确、尽可能多地提取有用信息仍是一个挑战。目前的轨迹信息主要包括病毒运动速度、运动模式、扩散系数、共定位情况等,如何更深入探究病毒运动轨迹与病毒感染进程以及病毒与细胞的相互作用关系,仍有待进一步的探索。
4.2 未来重点研究方向
针对目前存在的问题,未来应在以下几个方面开展重点研究。
1)完善病毒标记技术。继续开发光学性能更优良、尺寸更小的有机染料、荧光蛋白、标签蛋白、量子点和其他纳米粒子等,以减少病毒示踪过程中的荧光淬灭、细胞毒性等问题,为病毒标记提供更多选择。除了常用的荧光蛋白、荧光染料和量子点等,生物正交反应、核酸适配体等为病毒标记提供了新的思路。同时,优化现有标记方法,开发新的标记方法,建立更加特异、高效且尽可能降低对病毒活性影响的标记技术。这些都是目前制约病毒示踪技术应用的关键,也是亟待解决的问题,需要化学、物理和生物等交叉领域的研究者共同合作。
2)发展成像技术。目前,新的成像技术仍是亟需的。超分辨成像技术将光学显微镜的分辨率推进到了10 nm的量级,使研究者能在很高的空间分辨率下观察病毒感染过程。同时,研究者还可尝试将不同类型的显微镜联合应用,如STORM-AFM成像平台、常温超分辨光镜-常温电镜融合成像技术、冷冻超分辨光镜-冷冻电镜融合成像技术,以及包括超分辨光学STED成像模块、CARS成像模块和AFM成像模块等多模块联用的高分辨多功能化学成像系统。此外,发展活体成像仪器,建立可以应用于活体水平的单病毒示踪技术,为在体追踪病毒感染奠定基础。
3)优化图像处理技术。结合物理、数学和计算机的前沿技术,优化、发展图像分析算法,特别是利用计算机视觉和人工智能机器学习等前沿方法,提高算法的自动化和个性化程度,无需手动反复调试算法的相关参数,从而提高图像处理效率,降低手动分析产生的误差,以实现精确定位图像中的病毒粒子及重构其运动轨迹,提取具有相似特征的轨迹,对同一轨迹不同时刻的运动状态进行精细区分,从而便于研究者从成百上千条病毒运动轨迹中发现、总结规律,深入探究病毒侵染机制。
