导向定位测序数据的甲基化序列比对算法优化①
2022-01-06刘梦雅
刘梦雅, 徐 云
1(中国科学技术大学 计算机科学与技术学院, 合肥 230027)
2(安徽省高性能计算重点实验室, 合肥 230026)
DNA甲基化是指在DNA甲基化转移酶的作用下, 将甲基化基团选择性的添加到胞嘧啶(C)的过程.因此, 在人类基因组中一部分C被甲基化, 另一部分C未被甲基化, 在未改变基因序列的前提下, 控制基因表达[1,2].大量研究表明, 基因组中甲基化C的比例和所在区域, 能够为疾病的预测提供帮助, 同时也被证实在包括癌症在内的诸多疾病的治疗中发挥着重要的作用[3-6].
围绕全基因组甲基化的分析是近年研究的热点内容, 其中最重要的一步是将测序所产生的序列, 比对到参考基因组上, 获取整个基因组的甲基化状态[7].目前常用的测序技术是亚硫酸氢盐的全基因组甲基化测序,但由于此类测序技术需要用亚硫酸氢盐对原始DNA片段进行预处理, 降低了序列的复杂性, 增加了后续比对的难度.2019年出现的导向定位测序数据很好地解决这一问题, 其利用双端测序的优势: 一端是亚硫酸氢盐处理后的序列Read1, 另一端是原始序列Read2, 没有经过处理的原始序列更容易确定在参考基因组上的位置, 通过双端测序序列的位置关系, 实现对甲基化的精确检测[8].
然而, 现有导向定位测序数据(GPS)的比对方法先确定原始序列R2的20个候选比对位置, 时间消耗大; 之后再用动态规划算法确定甲基化序列Read1的比对位置, 算法本身的时间成本高, 且需对多个候选位置进行动态规划验证.同时, 根据Read2确定Read1的比对位置过于绝对, 可能会产生误判.现有亚硫酸氢盐测序(BS)中的比对方法能将70%-90%的序列确定到唯一的位置, 比对的准确率高达99%, GPS数据的现有比对方法, 相比之下仍有较大改进空间[9].
因此, 本文提出一种新的导向定位测序数据的比对算法.由于亚硫酸氢盐序列比对精度高达99%, 对于能确定唯一位置的甲基化序列不再用常规序列进行定位, 保证高精度的同时节约了时间.首先确定导向定位测序数据中的甲基化序列的候选比对位置; 然后根据甲基化序列和常规序列在参考基因组上对应的位置关系过滤偏离区域; 最后使用唯一比对序列的信息确定最佳比对位置.充分利用辅助信息, 实现以时间高效的方式将更多的甲基化序列比对到参考基因组上.
1 相关工作
目前, 对DNA甲基化进行检测的金标准是亚硫酸氢盐测序的全基因组甲基化测序, 随着导向定位测序数据的出现, 在实现对全基因组甲基化位点高度覆盖的同时, 带来了新的研究问题.接下来根据全基因组DNA甲基化测序数据的类型, 分别介绍数据的特点和相应比对方法, 分析其优缺点.
1.1 亚硫酸氢盐测序(BS)及其比对方法
亚硫酸氢盐测序技术通过对基因片段进行预处理,使得甲基化的胞嘧啶(C)保持不变, 未发生甲基化的C先转换成尿嘧啶(U), 再转换为胸腺嘧啶(T), 如图1所示[10].因此, 在DNA甲基化序列比对的过程中, 序列中的T有可能比对到参考基因组上的T或C, 但反之不行, 导致比对的难度增加[11].这是甲基化序列比对, 同常规DNA序列比对的不同之处.测序得到的基因序列, 称为BS-reads.甲基化分析中很重要的一步就是将BS-reads比对到参考基因组上, 确定其位置.
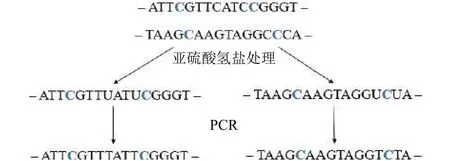
图1 亚硫酸氢盐测序过程
现有亚硫酸氢盐测序的比对方法分成两种, 分别是基于三字符集和基于通配符的比对方法.基于三字符集方法的特性, 是把BS-reads和参考基因组中的C都转化成T, 将问题转化成常规的DNA序列比对,在候选位置确定后, 再根据转化前的相似性对候选位置进行过滤, 代表方法有Bismark[12], GEMBS[13], BSSeeker3[14], BatMeth2[15].基于通配符方法的特性是BS-reads中的C转化成一个通配符, 同时允许通配符比对到参考基因组上的C和T, 代表方法有BSMAP[16]和RMAP[17].
随着越来越多的甲基化数据被测出, 这两类比对方法针对BS-reads不对称比对的特点, 适应序列长度短(40 bp-400 bp)、数量多、规模大的特性, 实现将甲基化序列快速比对到参考基因组上, 使得全基因组甲基化分析成为可能.但亚硫酸氢盐预处理将未发生甲基化的C转化成T, 在大部分序列比对中, 字符集从4字符集(A、T、C、G)变成了3字符集(A、T、G),降低了序列的复杂性, 增加了BS-reads唯一比对位置确定的难度, 同时使参考基因组中重复区域的甲基化状态分析更为艰难.
1.2 导向定位测序(GPS)及其比对方法
导向定位测序是一种新的全基因组DNA甲基化检测的方法.每条DNA链是由磷酸和脱氧核糖构成,3’端和5’端表示DNA链的两端, 其中连接磷酸基团的一端为5’端, 另一端是3’端.DNA的复制方向是从5’端到3’端.测序数据中3’端的序列保持不变, 5’端的未甲基化的C转化成T, 甲基化的C保持不变[8].获得的两条DNA序列(Read1和Read2), 其中Read1中未甲基化的C转化成T, 和亚硫酸氢盐测序方法处理后的序列特性一致; Read2是原始DNA序列, 更容易比对到参考基因组上, 如图2所示.在Read2比对到参考基因组之后, Read1比对到参考基因组的范围也相应确定.其中Read2对Read1位置的确定起到定位作用, 为后续全基因组甲基化的分析奠定了基础.
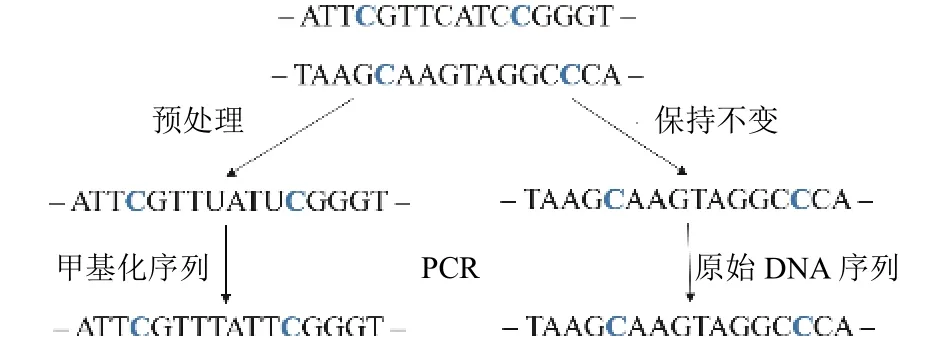
图2 导向定位测序过程
现有方法调用bowtie2[18]将Read2比对到参考基因组上, 获取Read2在参考基因组上的20个候选比对位置.由于Illumina测序原理可得, Read1位于Read2下游的相反链上, 且由于GPS测序库中的碎片大小是400 bp-500 bp, 可以确定Read2的比对范围.通过使用Smith-Waterman算法[19], 将Read1比对到Read2所在参考基因组下游1 kb的反链上, 获取Read1和参考基因组上局部相似性最高的位置.
新型测序数据的出现, 实现以较低的覆盖率(5X)获得甲基化序列, 降低了甲基化测序的成本, 检测甲基化没有序列偏好.同时, 比对过程中使用Smith-Waterman算法, 允许Read1中的T比对到参考基因组上的T或C, 以解决甲基化序列比对中C/T的不对称性比对问题.为受亚硫酸氢盐预处理影响较大的基因片段和部分物种, 提供了甲基化分析的新方法, 使得这部分序列甲基化信息的精确检测成为可能.但使用Smith-Waterman算法对多个候选比对位置进行动态规划验证, 需要大量的时间.且未考虑仅允许Read1中的C比对到参考基因组中的C, 有可能导致Read1的错误比对, 从而影响到后续全基因组甲基化的分析.且现有GPS数据的唯一比对比例为79.8%-82.3%, 仍有提升的空间.
2 比对算法设计和优化
本文首先将导向定位数据中的甲基化序列比对到参考基因组上, 随后利用和常规序列间的位置关系对候选位置进行过滤, 最后对仍不能确定位置的甲基化序列, 利用唯一比对位置的信息进行定位, 该方法主要包括4个步骤: (1)数据预处理; (2)定位候选位置;(3)过滤偏离区域; (4)确定最佳位置.
2.1 数据预处理
由于GPS库的建立, 需要用到T4 DNA聚合酶处理基因片段, 从而保证Read2中的序列和原始DNA片段一致, 最后获取双端测序序列(Read1和Read2).但T4 DNA聚合酶可能产生处理不足或过度处理的现象,直接影响获取数据的准确性, 影响比对的效率.所以,需要找到Read1和Read2处理的边界, 进而对数据进行预处理[8].
参考基因组中CH的甲基化水平较低, 若序列中出现CH, 则说明酶处理充分.Read2位于参考基因组的反链上, 根据碱基互补配对原理, 可知CH在Read2上的表现形式是[A/G/T]G.通过寻找[A/G/T]G确定酶处理边界, 对Read2进行预处理.如图3所示, 最靠近右端, 且满足要求的处理边界是TG.确定处理边界后,保留边界右边的序列作为处理后的Read2序列.
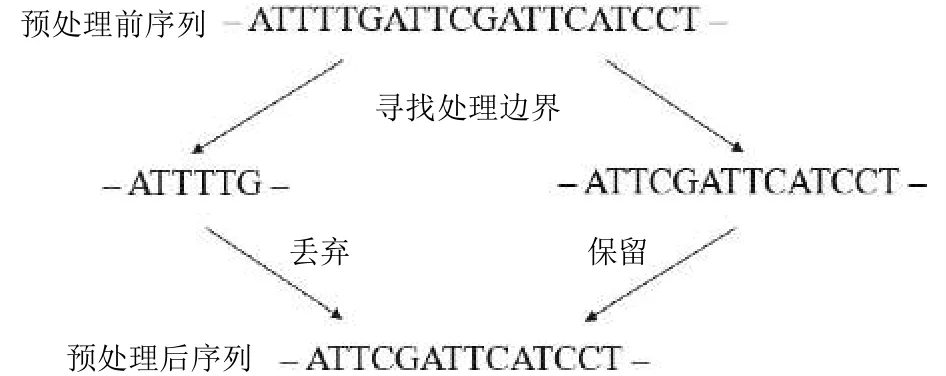
图3 数据处理示意图
2.2 定位候选位置
本文直接将甲基化序列比对到参考基因组上.一方面由于亚硫酸氢盐测序序列比对准确率较高, 另一方面易比对到多个位置的比例约为20%-30%, 直接比对甲基化序列在保证准确率的同时, 减少了后续的计算成本.本文使用基于三字符集方法和种子扩展策略的亚硫酸氢盐比对工具BitmapperBS[9]进行修改, 其包含高效的数据结构FM-tree, 针对数据三字符集特性对传统FM-index索引进行优化, 能够获得高达99.36%的准确率.
首先Read1比对到参考基因组后, 分成两部分.如图4所示, 将能够确定唯一位置的序列称为Unique Reads; 比对到多个位置的序列称为Multireads, 这部分序列比对到参考基因组的多个相似度较高的位置, 或者比对到了参考基因组的重复区域.
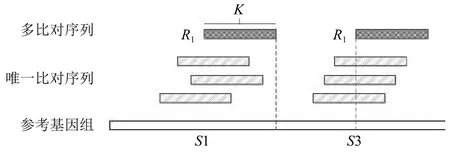
图4 唯一比对序列和多比对序列举例
后续处理主要针对Multireads, 找到其至多20个候选比对位置.将Multireads符号化表示为集合M, 设R1为集合M中的一条序列, 候选比对位置的个数为n,其候选比对位置集合P(R1)表示为:
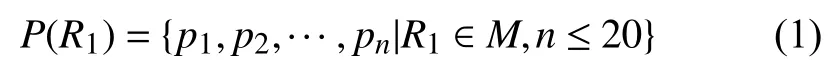
2.3 过滤偏离区域
针对GPS数据中的测序原理可得, Read1位于Read2下游的相反链上, 且距离相差不大于1000 bp.通过利用Read2的位置信息作为辅助信息, 对Read1的候选比对位置进行限制, 过滤位于偏离区域的候选位置.
设与R1相对应的另一端序列是R2, 首先使用bowtie2将R2比对到参考基因组上, 其候选比对的个数为m, 得到候选比对集合:

对Read1和Read2的候选位置进行两两比较, 过滤掉Read1候选比对集合中不能与Read2成对的位置.如图5所示,R1的候选比对位置集合P(R1)中只有p1和p3存在与之相对应的pos1和pos3, 所以对其余位置进行过滤, 此时P(R1)={p1,p3}.若此时R1的候选比对位置个数为1, 则转化为Unique Reads, 否则其仍在Multireads的集合M中.
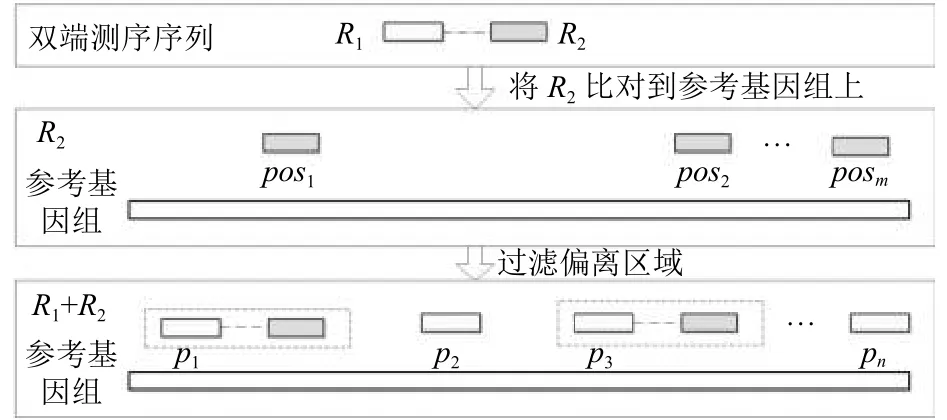
图5 过滤偏离区域
2.4 确定最佳位置
过滤偏离区域后, 使用与Multireads重叠的Unique Reads信息, 以及对应参考基因组之间的信息, 逐个碱基计算相应位置的可能性, 最后对候选集合中每个位置得到一个总的得分, 确定最有可能的比对位置.
设甲基化序列R1的长度为K, 比对到候选比对位置的概率S为:

其中,R1的第一个碱基比对到参考基因组对应位置的概率为s1, 依次类推得第K个碱基比对到参考基因组对应位置的概率为sK.如图6所示,s1-sK的计算使用工具BAM-ABS[20], 该工具使用贝叶斯模型, 以Multireads和参考基因组之间的错配信息和对应甲基化区域信息;以及重叠Unique Reads中获得的SNP和甲基化区域信息作为先验概率, 计算比对到每个位置的可能性.最后选取候选比对集合中得分最高的位置为最佳比对位置.
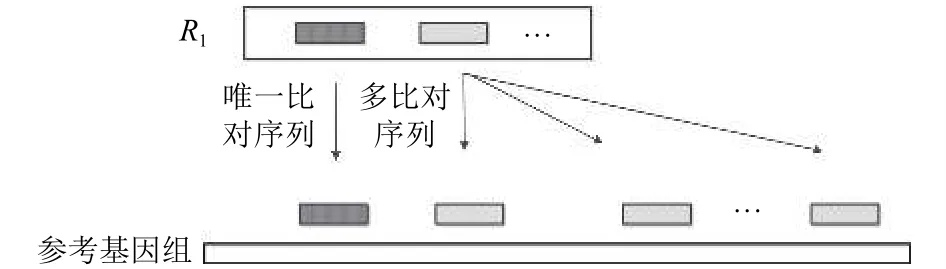
图6 找最佳位置的过程
3 实验分析
3.1 数据集和实验环境
本文分别在真实和模拟数据集中对两种方法进行比较, 真实数据集来自GSE92328, 在文献[8]中提出并被证实有利于甲基化信息的分析.本文使用其中的GPS数据SRR6443657和SRR6443658进行后续实验.模拟数据集使用模拟工具ART和Astair获得, 先用ART生成常规DNA数据, 再通过Astair对其中一条序列进行甲基化模拟.在未特殊声明时, 本文使用工具的默认参数进行比较.
本文的实验平台包括2个14核Intel Xeon Gold 5120处理器和512 GB内存, 操作系统为64位的Ubuntu 18.04.
3.2 评价指标
分别使用时间、唯一比对比率和准确率与现有方法进行比较.其中时间包括数据预处理和得到最终结果的时间, 建索引的时间不包括在内, 因为索引只需建造一次, 在后续实验中通用.
(1)唯一比对比率
该评价指标表示比对到唯一位置的甲基化序列占全部甲基化序列的比例.如式(4)所示,U表示唯一比对序列集合,n(U)表示唯一比对序列集合中序列的条数,N表示全部甲基化序列的条数.

(2)准确率
准确率这里表示唯一比对序列中, 比对到正确的位置所占的比例.如式(5)所示,n(R)是唯一比对集合U中比对到正确位置的序列个数.模拟数据集中序列在参考基因组上的位置是已知的, 当真实位置和比对结果相差200 bp以内, 则认为比对正确.真实数据集中序列在参考基因组上的位置是未知的, 故不进行准确率的验证.
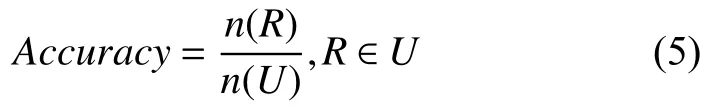
3.3 实验结果
分别使用模拟数据集和真实数据集探究本文方法和现有方法[8]的性能优劣.模拟数据集和真实数据的序列长度均为100 bp.数据规模分别为甲基化序列1w条、5w条、10w条, 常规DNA序列1w条、5w条、10w条.
(1)模拟数据集实验结果
如表1所示, 本文改进方法和现有方法相比, 准确率相差不大, 最多相差0.7%.而本文方法获得3-30倍时间性能的提升, 随着数据规模的增大, 对时间性能的提升越明显.同时本文方法获得6%-10%唯一比对比率的提升, 将更多的序列比对到唯一位置, 有利于后续甲基化信息的分析.因模拟数据集不能完全模拟真实数据中插入、删除, 以及发生测序错误、结构变异的情况, 更容易比对到参考基因组上, 唯一比对比率相比真实数据更高.
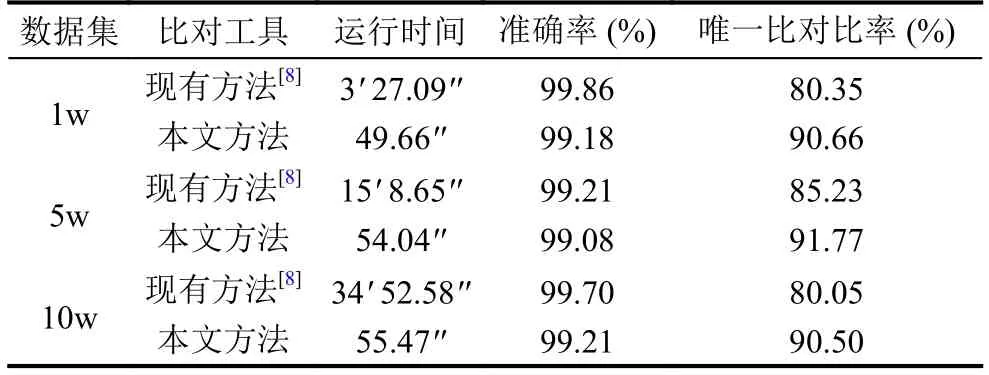
表1 模拟数据集实验结果
(2)真实数据集实验结果
通过实验探究了不同方法在运行时间、唯一比对比率方面的优劣.如表2所示, 在SRR6443657数据集中, GPS方法的运行时间从4 min到38 min, 受数据规模影响较大; 本文的改进方法在这3种数据规模下运行时间相差不大, 为56-67 s, 小数据集时比现有方法快约3倍, 大数据集时比现有方法快30倍, 对大规模数据集的提升效果更为明显.同时, GPS比对方法的唯一比对比率为79.32%-80.09%, 数据集规模对唯一比对比率的影响不大; 本文的改进方法唯一比对比率从85.37%到89.32%, 比之前方法提升了5%-10%, 且本文方法随着数据集规模越大, 唯一比对比率越来越大,因获取比对到唯一位置的序列信息越多, 更容易比对到唯一位置.第2个数据集整体结果和第1个数据集相似, 但唯一比对比率提升约为2%-6%, 较上一个数据集提升不明显.实验中发现部分甲基化序列未能找到与之配对的常规DNA序列, 使得该数据集比对难度增加.
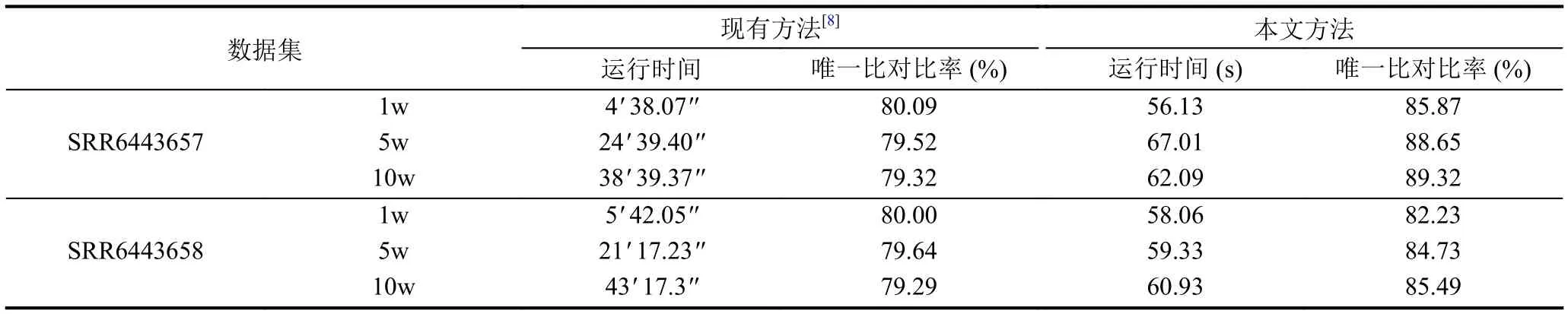
表2 真实数据集实验结果
4 结论与展望
本文提出了一种高效的导向定位测序数据的比对算法, 首先对数据进行预处理, 将甲基化序列定位到参考基因组上; 再利用双端测序中两端序列的位置关系,对甲基化序列的候选比对位置集合进行过滤; 最后通过比对到唯一位置的序列包含的信息, 找到最佳比对位置.实验结果表明, 本文方法能够加速比对过程, 将更多的甲基化序列比对到唯一位置, 且对大规模数据集的性能提升效果更为明显.下一步的研究工作是提出启发式的算法, 探究影响准确率的因素, 在比对精度上取得更好的效果, 并探究比对性能的提升对后续甲基化信息的影响.
