Newer variants of progressive familial intrahepatic cholestasis
2022-01-05VigneshVinayagamoorthyAnshuSrivastavaMoinakSenSarma
Vignesh Vinayagamoorthy, Anshu Srivastava, Moinak Sen Sarma
Vignesh Vinayagamoorthy, Anshu Srivastava, Moinak Sen Sarma, Department of Pediatric Gastroenterology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226014, Uttar Pradesh, India
Abstract Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of disorders characterized by defects in bile secretion and presentation with intrahepatic cholestasis in infancy or childhood.The most common types include PFIC 1 (deficiency of FIC1 protein, ATP8B1 gene mutation), PFIC 2 (bile salt export pump deficiency, ABCB11 gene mutation), and PFIC 3 (multidrug resistance protein-3 deficiency, ABCB4 gene mutation).Mutational analysis of subjects with normal gamma-glutamyl transferase cholestasis of unknown etiology has led to the identification of newer variants of PFIC, known as PFIC 4, 5, and MYO5B related (sometimes known as PFIC 6).PFIC 4 is caused by the loss of function of tight junction protein 2 (TJP2) and PFIC 5 is due to NR1H4 mutation causing Farnesoid X receptor deficiency.MYO5B gene mutation causes microvillous inclusion disease (MVID) and is also associated with isolated cholestasis.Children with TJP2 related cholestasis (PFIC-4) have a variable spectrum of presentation.Some have a self-limiting disease, while others have progressive liver disease with an increased risk of hepatocellular carcinoma.Hence, frequent surveillance for hepatocellular carcinoma is recommended from infancy.PFIC-5 patients usually have rapidly progressive liver disease with early onset coagulopathy, high alpha-fetoprotein and ultimately require a liver transplant.Subjects with MYO5 B-related disease can present with isolated cholestasis or cholestasis with intractable diarrhea (MVID).These children are at risk of worsening cholestasis post intestinal transplant (IT) for MVID, hence combined intestinal and liver transplant or IT with biliary diversion is preferred.Immunohistochemistry can differentiate most of the variants of PFIC but confirmation requires genetic analysis.
Key Words: Progressive familial intrahepatic cholestasis; Tight junction protein; Hepatocellular carcinoma; Biliary diversion; Microvillous inclusion disease
INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of intrahepatic cholestatic disorders caused by a defect in bile transport and secretion.It manifests in infancy or childhood and can progress to end-stage liver disease[1-3].Genetically confirmed PFIC accounts for 12%-13% of cholestatic disorders in infants and children[4].Disease variants are classified based on the specific bile transporter defects and all of them have an autosomal recessive inheritance.The three most prominent varieties are familial intrahepatic cholestasis-1, 2 and 3, which are caused by mutations in ATP8B1 gene encoding FIC1, ABCB11 gene encoding bile salt export pump, and ABCB4 gene encoding multidrug resistance protein-3 respectively (Figure 1).Nearly two-thirds of subjects with normal gamma-glutamyl transpeptidase (GGT) cholestasis (normally associated with PFIC except PFIC 3) do not have any mutations identified in ATP8B1 or ABCB11 genes[3].Detailed mutational analysis in patients with this phenotype has led to the identification of 3 more conditions, often known as PFIC 4, 5, and 6.PFIC 4 is caused by the loss of function of tight junction protein 2 (TJP2)[5], and PFIC 5 is due to NR1H4 mutation causing farnesoid X receptor (FXR) deficiency[6,7].MYO5B mutation, known to cause microvillous inclusion disease (MVID), is also reported to cause isolated cholestasis and is sometimes known as PFIC 6 though it is not yet recognized by the Online Mendelian Inheritance in Man[8].The exact incidence of newer variants of PFIC is not known due to the limited number of studies, which are mostly case reports or small case series.Based on the available literature, this review attempts to sensitize physicians to the disease.
GENETICS AND PATHOGENESIS
PFIC 4
TJP2 gene, located in chromosome 9q21 was first discovered in 1991 by Gumbineret al[9].It encodes a protein called tight junction protein 2 or zona occludens-2.Though named as tight junction protein, it is not present in the tight junction.Instead, TJP2 is a cytosolic protein, involved in maintaining cell-to-cell adhesion by linking the transmembrane tight junction proteins like claudin with the actin cytoskeleton.There are two types of claudini.e., claudin-1 (CLDN1) and claudin-2 (CLDN2), both of which are localized to the bile canalicular membrane[10].In TJP2 mutation, CLDN1 fails to localize to the bile canalicular membrane (Figure 2).This results in reduced integrity of the canalicular membrane and reflux of toxic bile acids through the paracellular spaces into hepatocytes, causing hepatocyte damage and cholestasis[11].TJP2 has a widespread expression, including the respiratory and central nervous systems.This may explain the systemic features reported in a few cases[11].The detergent action of the bile potentiates damage in the liver, which explains the predominant hepatic manifestations in this condition.

Figure 1 Pathogenesis of progressive familial intrahepatic cholestasis 1, 2 and 3.
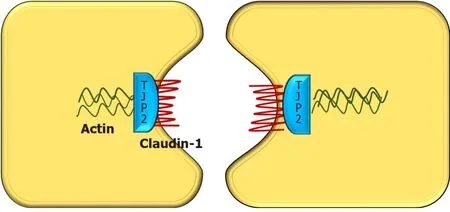
Figure 2 Diagrammatic representation of interaction between various tight junction proteins in hepatocytes.
PFIC 5
PFIC 5 is related to a deficiency of the FXR due to loss of function mutation in theNR1H4gene located in chromosome 12q23.NR1H4 related PFIC 5 is a less commonly reported variant, with < 10 cases reported by 2020.FXR, a protein translated from the NR1H4 gene was first described in 1995 by Formanet al[12].It belongs to a nuclear receptor group activated by farnesyl, an intermediate metabolite of the mevalonic acid synthesis pathway.FXR is the master regulator of cholesterol, bile acid, triglyceride and various sterol ring-containing compounds (Vitamin D, carotenoids, retinoids,etc.)[13].In the liver, the FXR acts as a nuclear bile acid-sensing receptor involved in the expression of bile salt export protein (BSEP) and sometimes MDR3[6,14].Apart from the liver, FXR is also expressed in the small intestine.Whenever bile acid levels are elevated in the ileal enterocytes, FXR is activated to induce the synthesis of fibroblast growth factor 19 (FGF19).FGF 19 is then transportedviaenterohepatic recirculation to the liver, where it binds to the fibroblast growth factor receptor 4/β-Klotho complex, and causes inhibition of bile acid synthesis by repressing CYP7A1.Elevated bile acid inside hepatocytes also activates FXR which induces ABCB11gene transcription, BSEP synthesis, and bile acid export from the liver.Hence, the NR1H4 mutation causes loss of BSEP expression, leading to the accumulation of toxic bile and hepatocellular damage (Figure 3).FXR is also involved in the regulation of coagulation factor synthesis by transactivating fibrinogen and kininogen genes.Thus, the FXR mutation leads to the development of vitamin K independent, early-onset coagulopathy, well before liver failure sets in[6].
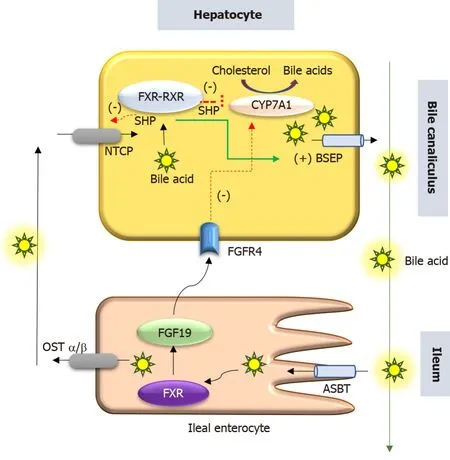
Figure 3 Schematic representation of role of farnesoid X receptor in hepatocyte.
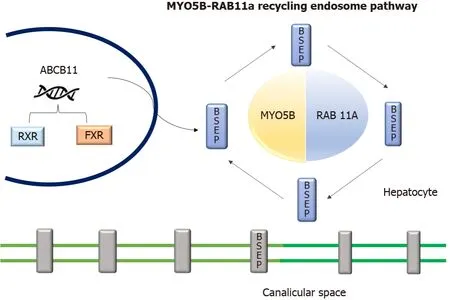
Figure 4 Diagrammatic representation of role of MYO5B and RAS-related GTP-binding protein 11A interaction and endosome recycling pathway and bile salt export pump expression.
Homozygous or compound heterozygous loss of function mutations (c.526C>T and c.419 420insAAA/intragenic 31.7-kb deletion, respectively) have been described[7].In one woman with intrahepatic cholestasis of pregnancy,NR1H4heterozygous variant (c.-1G>T) was found to be associated with cholestasis[15].
PFIC 6
TheMYO5Bgene located in chromosome 18q21.1 encodes an actin-associated molecular motor protein called MYO5B.MYO5B and RAS-related GTP-binding protein 11A (RAB11A) is essential for the epithelial cell polarization in multiple tissues (Figure 4).In hepatocytes, it is important for the localization of ATP-dependent bile canalicular transporters like BSEP to the canalicular membrane, and in the intestine, it is important for maintaining enterocyte polarity[16].MYO5B mutations disrupt the MYO5B/RAB11A recycling endosome pathway leading to defective targeting of BSEP[17].MYO5B gene mutations can result in cholestatic liver disease with or without associated MVID, which presents as intractable diarrhea in infancy[8,18].Staining of BSEP and MDR3 by immunohistochemistry in these patients is sub-canalicular in the location instead of the regular localization in the canalicular membrane[8].
There is a suggestion that the type of MYO5B mutation affects the clinical presentation[18,19].Less severe mutations have a loss of canalicular transporter function in hepatocytes without any loss of enterocytes functionality.These patients present with isolated cholestasis.In severe variants of mutations, there is a dysfunction of both bile canalicular transporter and enterocyte polarization.However, a severe loss of enterocyte function leads to a reduced bile acid absorption in the intestine and in turn decreased bile acid load to the hepatocyte, potentially preventing cholestatic manifestations[18].Patients with MVID more often have biallelic severe mutations in MYO5B.Biallelic mutations in the MYO5B-RAB11A interaction domain are more in MVID than those with isolated cholestasis[20].Thus, isolated cholestasis appears to reflect relatively mild MYO5B functional deficiency, whereas severe mutations in MYO5B primarily cause MVID[20].
CLINICAL PRESENTATION
Intrahepatic cholestasis is the hallmark of how these 3 genetic conditions present.Most often, patients present with variable combinations of pruritus, jaundice, pale stools, and failure to thrive.The published literature on each of these three entities (TJP2, FXR, and MYO5B) is limited and has been summarized in Tables 1-3 respectively.
PFIC 4
A varying spectrum of clinical presentation, ranging from mild anicteric illness, recurrent jaundice to severe progressive liver disease has been described[5,11].Incomplete penetrant, homozygous, missense mutations affecting both isoforms of TJP2 have been shown to cause familial hypercholanemia in the Amish population which manifests as a mild anicteric disease with pruritus and steatorrhea.In this condition, the binding of TJP2 to claudins is impaired[21].Milder mutations of TJP2 are also known to be associated with intrahepatic cholestasis of pregnancy[15].
In the 12 cases reported by Sambrottaet al[5], 9 (75%) required liver transplantation (LT) while 2 had portal hypertension.In contrast, none of the 7 cases reported by Zhanget al[22] required LT, and cholestasis responded to medical therapy in a majority.Zhanget al[22] also showed that truncating or canonical splice-site biallelic TJP2 mutations caused a more severe presentation due to a complete loss of protein expression.In their study, 3 children with severe mutations had growth failure.While the other 3 cases with missense variants had normal growth and sustained response in pruritus with ursodeoxycholic acid (UDCA) and cholestyramine.
All homozygous mutations are predicted to abolish protein translation and a complete loss of function[5].Mutations involving missense and frame deletion lead to less severe clinical disease due to residual TJP2 protein expression[22].This suggests the presence of a genotype-phenotype correlation based on the amount of remnant functional TJP2 activity.
There is a higher risk of developing hepatocellular carcinoma (HCC) in these cases, similar to that seen in PFIC 2 patients.Subjects can either present with a spaceoccupying lesion (SOL) in the liver or are detected to have HCC after LT on histology of the explanted liver[23,24].This predisposition to HCC highlights the importance of close follow-up and regular monitoring.
PFIC 5
FXR is the master player of bile acid regulation and plays an important role in reducing bile acid-induced hepatotoxicity.Rapidly progressive liver disease and early onset vitamin K independent coagulopathy are the main features of this condition.The details of the 8 published cases are given in Table 2.A majority of patients presented early in the first 3 mo of life and progressed rapidly to liver failure.Patients have markedly increased alpha-fetoprotein and deranged international normalized ratio.Without a liver transplant, 5/8 died in infancy itself.Three cases survived post-liver transplant, of which 2 were found to have liver function abnormality with graft steatosis in the follow-up[6].This post-transplant hepatic damage may be attributed to the altered enterohepatic circulation and FXR signalling in these cases.The absence of FXR in the intestine leads to low FGF 19 levels and this allows for continued and increased synthesis of bile acids by the liver[25].Intrahepatic cholestasis of pregnancy has been reported and attributed to the downregulation of BSEP in this condition[26].

Table 1 Clinical characteristics and outcome in patients with TJP2 mutation
PFIC associated with MYO5B defects
Patients with MYO5B mutations can present with isolated cholestasis, isolated MVID, or both MVID and cholestasis.Typically, the child presents with jaundice, pruritus, and hepatomegaly.In patients with MVID and cholestasis, the onset of cholestasis may be pre or post-small bowel transplant.The exact explanation as to why some MVID cases develop cholestasis while others do not is unclear but it may be related to the severity of mutation (vide supra).The summary of the clinical presentation of 29 cases with MYO5B mutation, as reported in 4 papers, is shown in Table 3.Even in siblings with the same mutation and presentation with cholestasis, the disease severity may vary[20].This suggests the possible role of modifier genes or environmental factors.Among Han Chinese children, defects in MYO5B accounted for approximately 20% of cases of idiopathic low-normal GGT intrahepatic cholestasis[20].
INVESTIGATIONS AND THE APPROACH TO DIAGNOSIS
The main steps for making a diagnosis of PFIC and determining the specific type in any given child with cholestasis are as follows: Step 1: Detailed history and physical examination including family history, consanguinity, extraintestinal symptoms, growth, nutritional deficiencies, and features of advanced liver disease; Step 2:Complete liver function test with GGT.Low-normal GGT is seen in ATP8B1, ABCB11, TJP2, NR1H4, and MYO5B disease.Early-onset of vitamin K unresponsive coagulopathy is a feature of NR1H4 disease; Step 3: Radiologic imaging.Ultrasonography (USG) of the abdomen is useful to exclude structural causes of neonatal cholestasis, like biliary atresia or choledochal cyst.The presence of biliary radicle dilatation may suggest sclerosing cholangitis, which needs to be confirmed by MRCP.USG is also useful to document features of advanced liver disease like ascites, splenomegaly, dilated portal vein, and collaterals.Gall stones have been reported in TJP2 disease, as also in PFIC 2 and 3.The presence of hepatic SOL raises suspicion of HCC and needs evaluation by triple-phase CT and alpha-fetoprotein.Early HCC is a feature of TJP2 disease; Step 4: Liver histology including immunohistochemistry and nextgeneration sequencing (NGS).Liver biopsy shows canalicular cholestasis in all threetypes (TJP2, NR1H4, and MYO5B defects) along with a variable degree of fibrosis and giant cell transformation[6].On electron microscopy, the tight junctions appear elongated and lack the densest part of the zona occludens in PFIC 4[6].In subjects with MYO5B and liver disease, electron microscopy will show dilatation of the bile canalicular lumen, canalicular thickening, and disappearance of the microvilli apart from cholestasis[17].Inclusion bodies are not seen in the hepatocytes on transmission electron microscopy, in contrast to the findings in intestines in MVID[17].The comparative features at histology in these three types are given in Table 4.
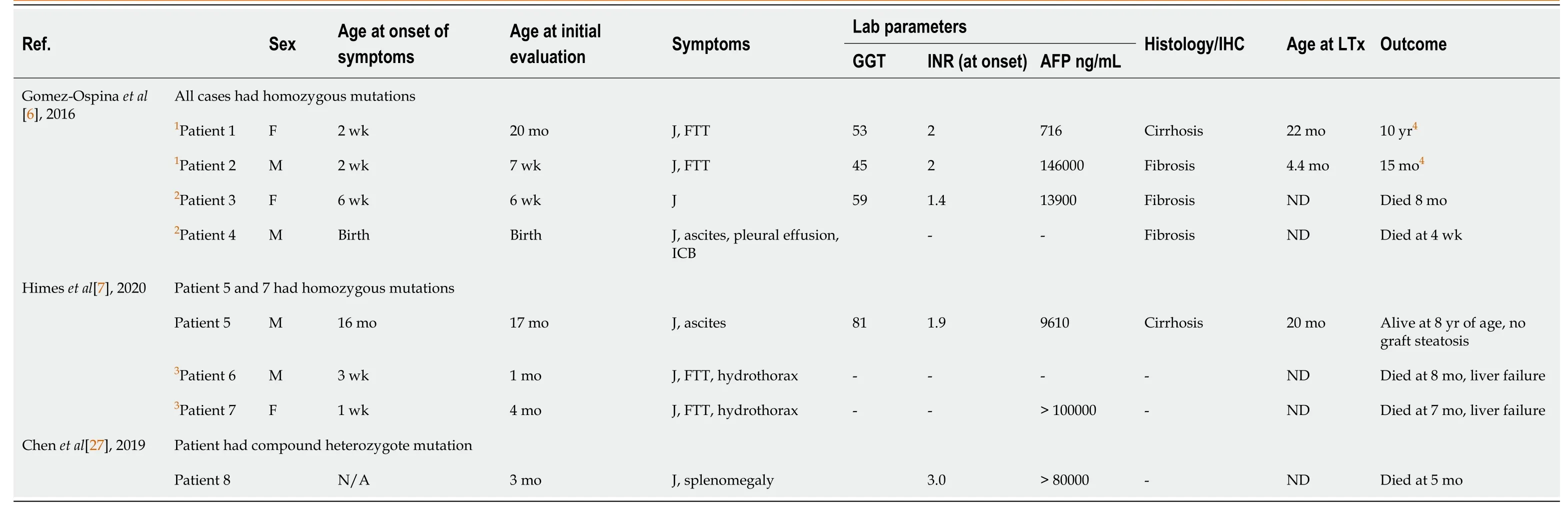
Table 2 Clinical characteristics and outcome in patients with NR1H4 mutation

Table 3 MYO5B mutation clinical characteristics and outcome
A complete panel of immunohistochemistry including BSEP, MDR3, TJP2, FXR, MYO5B, and Claudin1 can help in identifying the subtype of PFIC as shown in Table 4.However, simultaneous NGS for multiple genes (cholestasis panel) is a rapid and affordable way of confirming the molecular diagnosis[27].A recent study has shown that a molecular genetic diagnosis can be made in a quarter of cases with neonatal cholestasis using NGS[28].A study with a 66-gene cholestasis panel in 2171 cholestatic children and young adults, had a diagnostic yield of 12% and turnaround time of only 21 d[29].The simultaneous testing for multiple genes helps in not only confirming the diagnosis but also in excluding other conditions.NGS is becoming the test of choice in the primary evaluation of patients with PFIC phenotype as it is noninvasive in comparison to liver biopsy and immunohistochemistry.For cases in whichthe panel yields a negative result and the index of suspicion is high, further testing by the whole exome (WES) or whole-genome (WGS) sequencing may be done.The presence of variables of unknown significance and monoallelic pathogenic/likely pathogenic variants in a significant proportion of cases highlights the complexity of analysis and the need for expertise for proper interpretation.Also, the ongoing discovery of new genes requires expansion of the genetic testing panel from time to time.

Table 4 Comparison of clinical features, laboratory profile and outcome in progressive familial intrahepatic cholestasis 4, 5 and 6
DIFFERENTIAL DIAGNOSIS
The main differentials to be considered in a patient with intrahepatic cholestasis with low-normal GGT (< 100 U/L) include bile acid synthetic defect (BASD), arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome, and USP53 related cholestasis, apart from the different types of PFIC (1, 2, 4, 5 and MYO5B associated).Type 3 PFIC (ABCB4) has raised GGT[1,30].The serum bile acids are raised in PFIC and ARC syndrome, while they are low in the BASD.These entities can be differentiated by their distinct clinical presentation and liver histopathology with immunohistochemistry.However, the confirmation of diagnoses is best done by genetic analysis.
Bile acid synthetic defects
In bile acid synthetic defects (BASD) there is an accumulation of toxic bile acid intermediates in the hepatocytes due to deficiency of various enzymes involved in bile acid synthesis.Patients present with cholestatic jaundice, overt steatorrhea and florid manifestations of fat-soluble vitamin deficiencies like rickets.Pruritus is distinctly uncommon.Sometimes they may also present with neonatal liver failure, and cholestatic liver disease along with neurological manifestations like hypotonia, seizures[31].BASD is diagnosed with fast-atom bombardment mass spectrometry of urine, which shows the accumulation of the distinct bile acid intermediaries due to a block in the bile acid synthesis pathway.Genetic analysis is confirmatory.Supplementation of cholic acid (CA) and chenodeoxycholic acid (CDCA) along with fatsoluble vitamins is the mainstay of therapy[32].
ARC syndrome
ARC syndrome (MIM 208085) is a rare multisystem disorder with autosomal recessive inheritance.It includes a triad of arthrogryposis, renal tubular acidosis, and neonatal cholestatic jaundice.Some patients may have accompanying features like ichthyosis (approximately 50%), platelet anomalies (approximately 25%), agenesis of the corpus callosum (> 20%), congenital cardiovascular anomalies (approximately 10%), and deafness.The clinical features are very useful to suspect the diagnosis, which is confirmed by a demonstration of mutations in the VPS33B or VIPAR gene.Histopathology shows bile duct paucity, giant cell transformation, bile plugs, and portal fibrosis.Caution is required before proceeding with a renal or liver biopsy due to the increased risk of a life-threatening bleed.Treatment is supportive and includes management of joint contractures, renal tubular acidosis, and cholestasis (UDCA, fatsoluble vitamins)[33].
USP53 related cholestasis
USP53 encodes an enzyme known as ubiquitin carboxyl-terminal hydrolase 53, which belongs to the de-ubiquitinating enzyme family and helps in maintaining cell integrity by interacting with TJP2 in hepatocytes.Whole-exome sequencing in 69 Han Chinese infants, with low GGT cholestasis without any pathological variants in ATP8B1, ABCB11, NR1H4, TJP2, and MYO5B genes, showed the presence of biallelicUSP53mutations (homozygous or compound heterozygous) in 7 patients[34].All these children had cholestatic jaundice in infancy and responded to medications (UDCA, cholestyramine).Liver biopsy showed varying levels of lobular disarray and hepatocellular and canalicular cholestasis, rosetting, portal tract fibrosis, ductular proliferation, and giant-cell transformation.Ultrastructural examination in 2 cases revealed abnormality of tight junction complexes and expression of TJP2 and CLDN1 were reduced.Two children also had sensorineural hearing loss.In another report on the novel USP53 mutation, three members from the same family (2 sisters and a cousin) had low-GGT cholestasis, pruritus, elevated transaminases, very high alkaline phosphatase, and sensorineural hearing loss (n= 2).One of them required LT because of intractable pruritus[35].
Alagille syndrome
Alagille syndrome is also known as arteriohepatic dysplasia or syndromic paucity of interlobular bile ducts.This disorder is autosomal dominant with variable phenotypic penetrance.Alagille syndrome is one of the commonest causes of genetic cholestasis[36].The defining feature is cholestasis with multisystemic involvement.Features include neonatal cholestasis in 95%, extrahepatic biliary hypoplasia, pruritus, xanthoma and associated facial dysmorphism.Structural cardiac defects such as peripheral pulmonary stenosis and septal defects are seen in 88%.Vertebral anomalies, ocular abnormalities most commonly posterior embryotoxon, renal dysplasia, vascular anomalies like Moyamoya disease, carotid and subclavian artery aneurysm are the other systemic features.Genetic analysis revealsJAG1mutation in the majority (approximately 90%) andNOTCH2mutation in minority[35].
Citrin deficiency
It is caused due to SLC25A13 (Solute Carrier family 25) gene mutation located in chromosome 7q21.3.The disease spectrum includes neonatal intrahepatic cholestasis, failure to thrive and dyslipidemia, and adult-onset type II citrullinemia.Chubby cheeks in infancy are a hallmark finding.These children also have a characteristic history of aversion to carbohydrates and a dietary preference towards a protein and lipid-rich diet[37].
Neonatal ichthyosis-sclerosing cholangitis syndrome
Neonatal Ichthyosis Sclerosing cholangitis is a rare cause of neonatal cholestasis with an autosomal recessive inheritance pattern.It is caused due to a mutation in the CLDN1 gene which encodes the CLDN1 protein located at the tight junction.This condition presents with neonatal cholestasis, cicatricial alopecia, ichthyosis and pruritus.Magnetic resonance cholangiopancreatography will show features of sclerosing cholangitis[38].
Other PFIC subtypes
Amongst the different PFIC subtypes, PFIC 1, 2, 4, 5 and 6 have low-normal GGT cholestasis.The presence of diarrhea is a feature of PFIC 1 and MYO5B disease.While neurological symptoms may be seen in ARC syndrome and sometimes in patients with MVID, a higher risk of HCC is a feature of TJP2 and BSEP deficiency.Table 4 gives the comparison of the clinical features and investigations in TJP2, FXR, and MYO5B defects.A detailed description and comparison of PFIC 1 and 2 are given elsewhere[39].
TREATMENT
Medical management
The main components include counselling of parents in detail, providing adequate nutrition, correcting vitamin deficiencies, controlling pruritus, managing complications like ascites, variceal bleedingetc., growth monitoring, and vaccination[40].
Nutritional therapy:A diet that provides adequate calories (125%-140% of RDA) and protein (2-3 g/kg) with supplementation of medium-chain triglyceride and fat-soluble vitamins is recommended[41].The doses of vitamin supplementation may need modification based on clinical signs and symptoms of vitamin deficiency and serum level monitoring (if available).Anemia, if present, needs to be corrected.Ageappropriate immunization including vaccination against hepatotropic viruses (hepatitis A and hepatitis B) is essential.
Management of pruritus:Pruritus is one of the most disabling symptoms in these children.Apart from skincare, medications such as UDCA, cholestyramine, rifampicin, naltrexone, and sertraline are used for controlling pruritus.These aspects have been addressed in detail elsewhere[42].There are no published reports on the use of FXR agonists like obeticholic acid, or apical sodium–bile acid transporter inhibitors like maralixibat in PFIC 4, 5 and MYO5B related diseases.Long-term follow-up includes growth monitoring, monitoring for nutritional deficiencies, and HCC surveillance, especially in TJP2 related cholestasis.
Biliary diversion
Biliary diversion (BD) takes away bile from the intestine, thereby reducing the reabsorption of bile acids through the enterohepatic circulation[43].It has an important role in the alleviation of pruritus that is refractory to medical management in PFIC 1 and 2[44].The role of BD is not well known in the newer variants of PFIC.BD has been tried in MVID patients who developed worsened cholestasis post intestinal transplant and was found to be helpful[17].In MYO5B mutation, the ongoing cholestatic liver disease worsens after the intestinal transplant, leading to progressive liver fibrosis.Hence combined liver and intestinal transplantation are preferred.But in cases of isolated intestinal transplants, gallbladders should be preserved so that in case the cholestasis worsens, partial external biliary drainage can be attempted.The ileal bypass should be avoided as it removes a part of the transplanted bowel and doesn’t result in long-term remission of cholestasis[16].
Liver transplant
LT is to be considered in children with decompensated chronic liver disease, growth failure (not amenable to dietary modification), refractory pruritus, or associated complications like hepato-pulmonary syndrome.In NR1H4 related PFIC, an early transplant may be required due to progressive liver disease with decompensation.Post liver transplant graft steatosis may develop in patients with NR1H4 mutationassociated cholestasis[6].
Genetic counseling
Once a child is confirmed to have PFIC, parents need to be counselled about the nature of the disease and the autosomal recessive pattern of inheritance.A geneticist should be involved in counselling about future pregnancies and testing during pregnancy[45].
CONCLUSION
TJP2, FXR, and MYO5B are recent additions to the three well-known types of PFIC (1, 2, and 3).This review has described the genetics, clinical profile, investigative findings, and treatments of these newer entities.There are gaps in our understanding of these conditions due to the limited literature at present.Advances in bioinformatics and techniques of next-generation gene-sequencing will help us study the genotypephenotype correlation and synergistic effect of multiple mutations.Despite the recognition of these entities, not all cases with the PFIC phenotype have a confirmed genetic diagnosis, which indicates the presence of other causative genes that are waiting to be discovered.
杂志排行

World Journal of Hepatology的其它文章
- Non-alcoholic fatty liver disease in irritable bowel syndrome: More than a coincidence?
- Liver-side of inflammatory bowel diseases: Hepatobiliary and druginduced disorders
- Gastrointestinal and hepatic side effects of potential treatment for COVID-19 and vaccination in patients with chronic liver diseases
- Genotype E: The neglected genotype of hepatitis B virus
- One stop shop approach for the diagnosis of liver hemangioma
- Liver function in COVID-19 infection