一种低温热膨胀微球的制备
2022-01-04苏兰辉柴于登吴宇鹏
苏兰辉,柴于登,吴宇鹏
(浙江衢州巨塑化工有限公司,浙江 衢州 324004)
热膨胀微球(又称热塑性微球、发泡微球)由发泡剂(液态低沸点烷烃或其他化合物)和热塑性聚合物外壳构成[1-2],是一种具有壳核结构的聚合物微球。加热时,发泡剂汽化产生内压,热塑性聚合物外壳受热软化,微球向外膨胀;冷却后,微球仍能保持发泡形状[3-4]。微球发泡后,其直径可增至3~5倍,密度约从1 100 kg/m3下降至30 kg/m3[5-7]。由于其质轻和均一发泡的特性,膨胀后的微球在隔热、隔声、减重、减震等方面具有广泛应用,其研究也受到广泛关注[8]。
热膨胀微球按起始发泡温度范围可分为3类:低温(低于120 ℃)、中温(120~180 ℃)、高温(高于180 ℃)。其中,低温热膨胀微球通常用于低阻燃、低加工温度的材料改性,如用于皮革、油墨、印花、纸张改性等领域[9-10]。在这些领域,材料生产及加工温度过高将导致基材本身性能下降[11]。
热膨胀微球的制备主要使用悬浮聚合方式,分为有限凝结聚合、界面活化稳定聚合、界面活化聚合(微悬浮聚合)[12]。有限凝结聚合使用无机分散剂和低分子质量的脂肪酸和醇胺缩合物促进剂等分散剂作为分散体系,其优点是:低转速合成小粒径树脂,其粒径分布小、大小均一且不聚并。界面活化稳定聚合可以无机分散剂或有机分散剂作为分散体系,同时引入少量的表面活性剂辅助分散。界面活化稳定聚合的优点是可以合成小粒径;缺点是靠高转速剪切分散合成小粒径树脂,颗粒分布宽且易聚并。界面活化聚合(微悬浮聚合)使用表面活性剂作为分散体系,可以合成2 μm以下聚合物微球[13]。本文中采用有限凝结聚合法,通过调节单体比例、发泡剂用量、交联剂种类及用量,合成一种低温高膨胀倍率的热膨胀微球。
1 试验部分
1.1 试验原料和设备仪器
(1)原料。偏氯乙烯(VDC),自制,工业级;丙烯腈(AN),丙烯酸异丁酯(BA),浙江卫星石化股份有限公司,工业级;异戊烷、聚乙烯吡咯烷酮(PVP),国药集团化学试剂有限公司,分析纯;硅溶胶(50%),诺力昂新材料(苏州)有限公司,工业级;二甲基丙烯酸己二醇酯(HDDMA),上海阿拉丁生化科技股份有限公司,分析纯;过氧化月桂酰(LPO),上海阿拉丁生化科技股份有限公司,分析纯;去离子水,自制。
(2)设备仪器。1 L高压玻璃反应釜,CF-41循环式油温机,HRT-500K显微镜热台,SSD-550体视光学显微镜,JSM-5610LV扫描电子显微镜(SEM),Mastersizer-v3.62激光粒度分析仪,TMA-Q400热机械分析仪。
1.2 试验步骤
取一定量去离子水、硅溶胶、PVP、亚硝酸钠、氯化钠加入烧杯,开启搅拌,调节pH值至3~4,形成水相。取一定量VDC、AN、BA、发泡剂、过氧化二月桂酰、交联剂加入烧杯,开启搅拌,配制成油相。将水相和油相加入均化器,在8 000~10 000 r/min转速下均化5 min后,加入1 L反应釜,控制反应温度62 ℃,转速400 r/min,反应24 h。反应结束,降温出料,洗涤干燥。
1.3 测试方法
1.3.1 粒径及粒径分布
取热膨胀微球,加入去离子水,超声振荡5 min,配制成质量分数2%的悬浮液。滴加悬浮液至Mastersizer-v3.62激光粒度分析仪,进行测试。粒径分布宽度的表示方法采用span法。
1.3.2 微观形貌
将待测热膨胀微球置于导电胶,样品喷金后进行SEM表征。
取一定量发泡树脂置于显微镜热台,设置热台升温速率20 ℃/min,从20 ℃升至300 ℃,观察热膨胀微球发泡情况。
1.3.3 发泡性能测试
使用TMA-Q400热机械分析仪表征微球发泡性能,称取4 mg热膨胀微球置于铝制小坩埚内,测量样品高度,记为D样品。施加0.01 N载荷,空气氛围下以15 ℃/min升温速率至300 ℃,观察探针位置随温度的变化,Tstart为微球起始发泡的温度,Tmax为微球膨胀至最大时的温度。Dmax为微球最大膨胀高度,微球膨胀倍率=(Dmax+D样品)/D样品。
2 结果与讨论
2.1 软单体用量的影响
软单体的玻璃化转变温度较低。加入软单体可以降低聚合物外壳软化点,制备出发泡温度低、韧性优异的热膨胀微球。本研究主要采用BA作为软单体,研究其用量(BA质量/单体总质量)对热膨胀微球发泡性能和粒径的影响,结果如图1、图2所示。
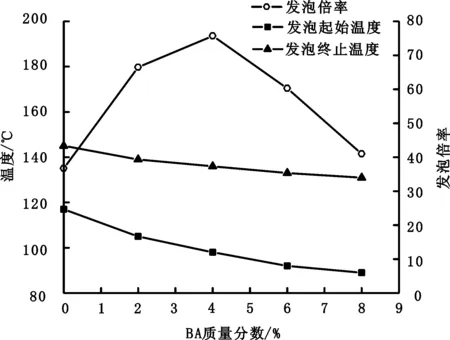
图1 BA单体比例对发泡性能的影响Fig.1 Effect of monomer BA content on foaming performance
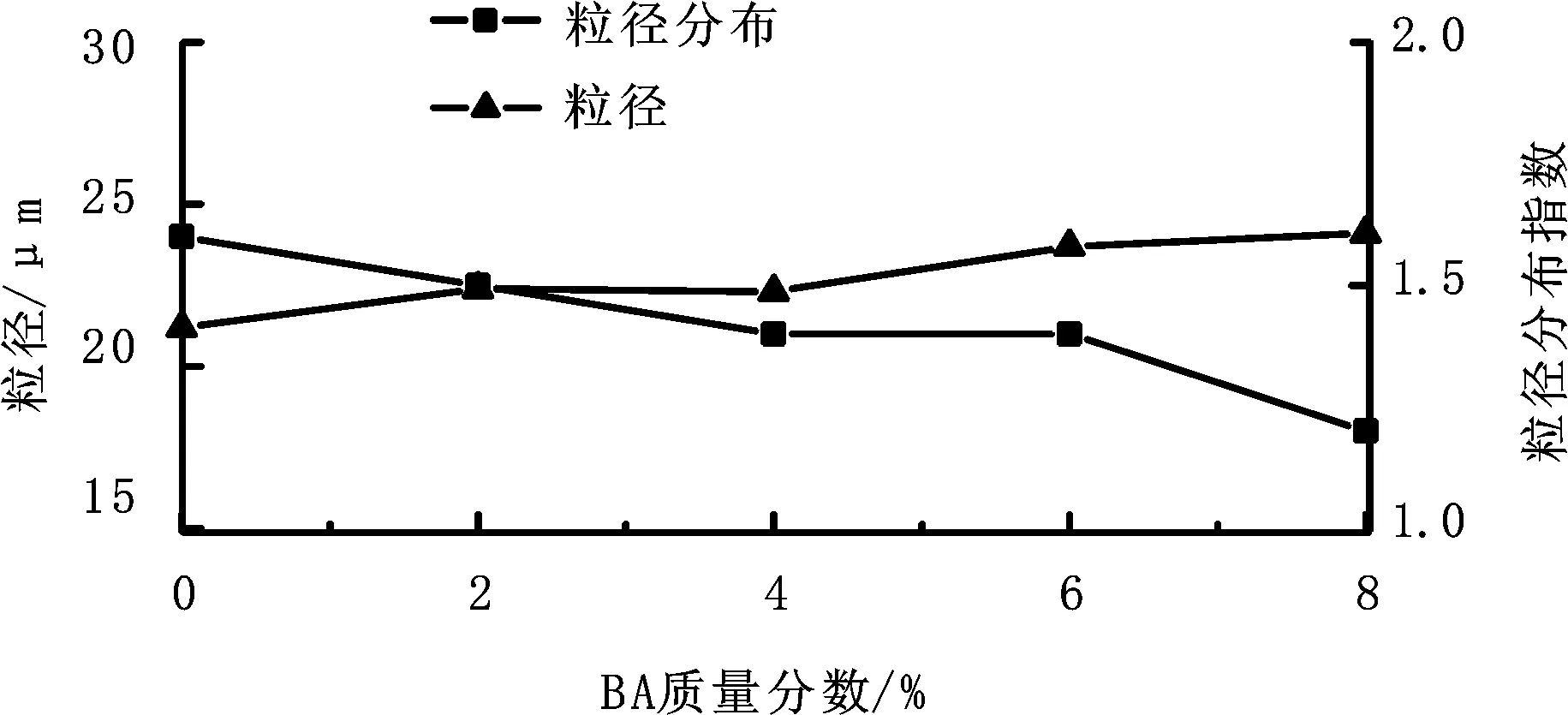
图2 BA单体比例对粒径及粒径分布的影响Fig.2 Effect of monomer BA content on particle size and its distribution
在图1中,随着BA单体比例增大,微球发泡起始温度和发泡终止温度开始降低,稳泡温程增大,发泡倍率则先增大后减小。当BA单体比例为4%时,热膨胀微球起始发泡温度为98 ℃,终止发泡温度为136 ℃,稳泡温程为38 ℃,发泡倍率为75.7%。随着BA单体增加,发泡起始温度和终止温度出现降低,是因为BA单体玻璃化转变温度低,随着其添加量增加,聚合物外壳软化点降低,微球发泡起始温度和终止温度降低。发泡倍率先增大后减小,则是因为BA加入量较小时,随着BA含量增加,聚合物外壳韧性增强,微球不易破损,微球发泡倍率增大。而当BA含量过大时,随着BA含量增加,聚合物外壳气密性下降,微球发泡倍率减小。
对于微球粒径,如图2所示,BA单体的加入,对微球粒径及粒径分布影响较小。从图2也可看出微球粒径分布较窄,有利于其发泡均一性。
2.2 发泡剂种类和含量的影响
发泡剂位于核壳结构内部,加热时发生汽化,从而给微球足够的内压使其外壳伸展体积膨胀。发泡剂须与聚合单体互溶而不溶于生成的聚合物。此外,发泡剂对聚合物外壳要有较低的透过性,且发泡剂不参与化学反应。本研究主要采用异戊烷作为发泡剂。研究发泡剂用量(异戊烷质量/单体总质量)对热膨胀微球发泡性能和粒径的影响,结果如图3、图4所示。
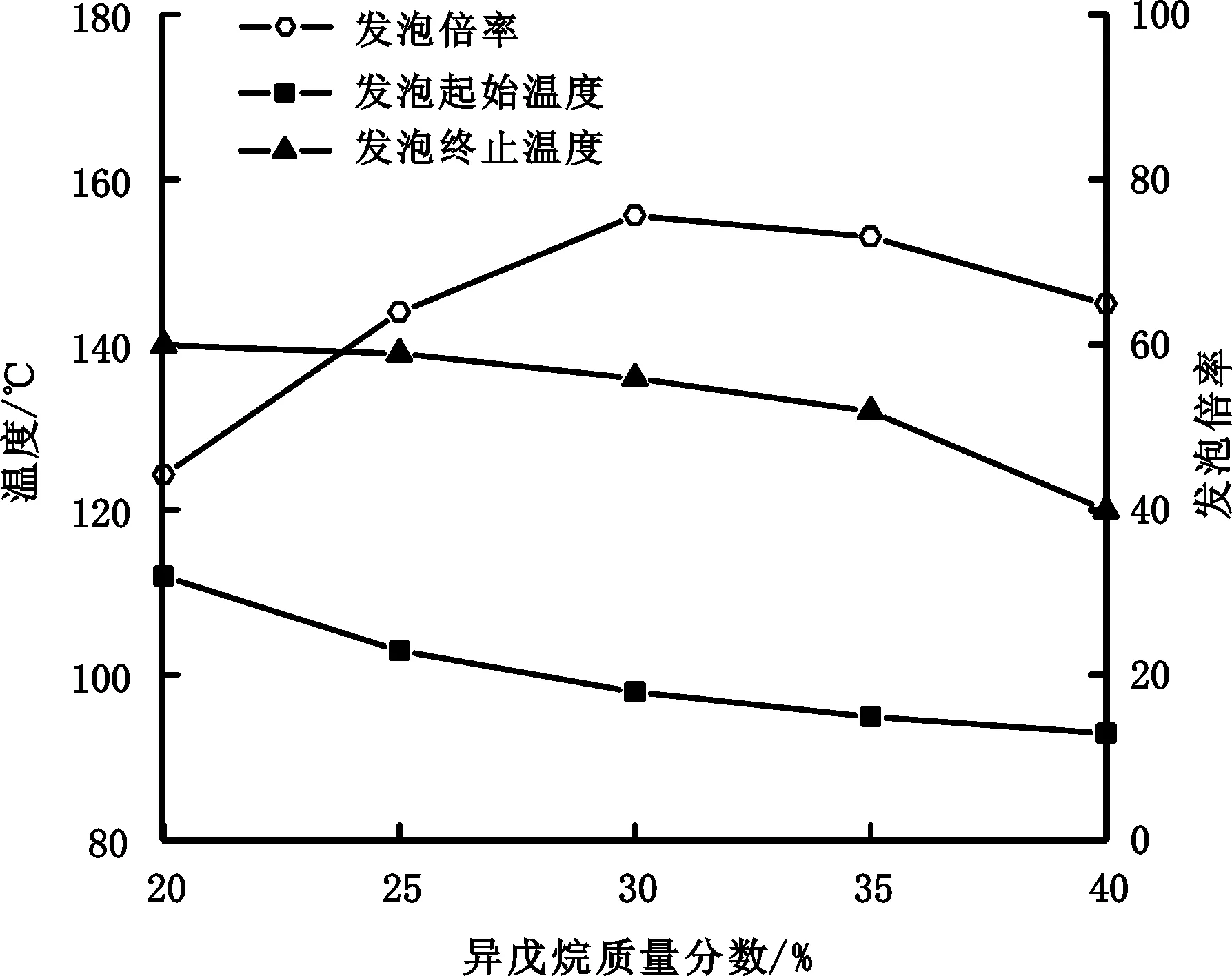
图3 异戊烷用量对微球发泡性能的影响Fig.3 Effect of isopentane added amount on the foaming properties of microspheres
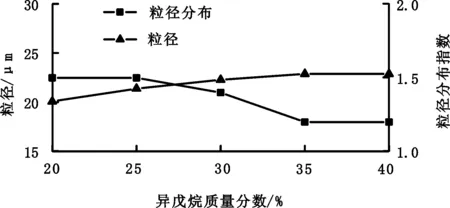
图4 异戊烷用量对微球粒径及粒径分布的影响Fig.4 Effect of isopentane added amount on particle size and its distribution
图3中,随着发泡剂用量增加,微球发泡起始温度和发泡终止温度降低,稳泡温程和发泡倍率则先增大后减小。当异戊烷用量为30%时,热膨胀微球发泡性能最佳。随着异戊烷用量增加,发泡起始温度和终止温度出现降低,是因为随着异戊烷用量增加,微球在更低的温度下就能产生足够的内压使其外壳伸展,体积膨胀,同时也在更低的温度下膨胀微球出现破泡现象。发泡倍率先增大后减小,则是因为在适当的发泡剂用量下,增大发泡剂用量,发泡剂汽化时可以形成更大的膨胀微球。而当发泡剂过量时,膨胀微球较易出现破损漏气,导致发泡倍率下降。
对于微球粒径,如图4所示,发泡剂用量增加,微球粒径增大。因为聚合物外壳中单体聚合时体积产生收缩,而发泡剂含量高时,聚合物外壳更薄,体积收缩更不明显,所以微球具有更大粒径。从图4也可以看出,微球粒径分布较窄,这有利于其发泡均一性。
2.3 微观形貌
取4%BA、30%异戊烷合成的热膨胀微球,采用SEM观察膨胀前发泡微球(如图5所示)。取热膨胀微球,置于显微镜热台上,使用光学显微镜观察发泡过程,结果如图6所示。

(a)低倍

(b)高倍图5 低温热可膨胀微球SEM图Fig.5 SEM of low-temperature thermal expansion microspheres
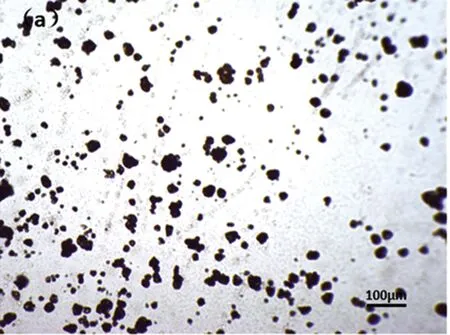
(a)发泡前
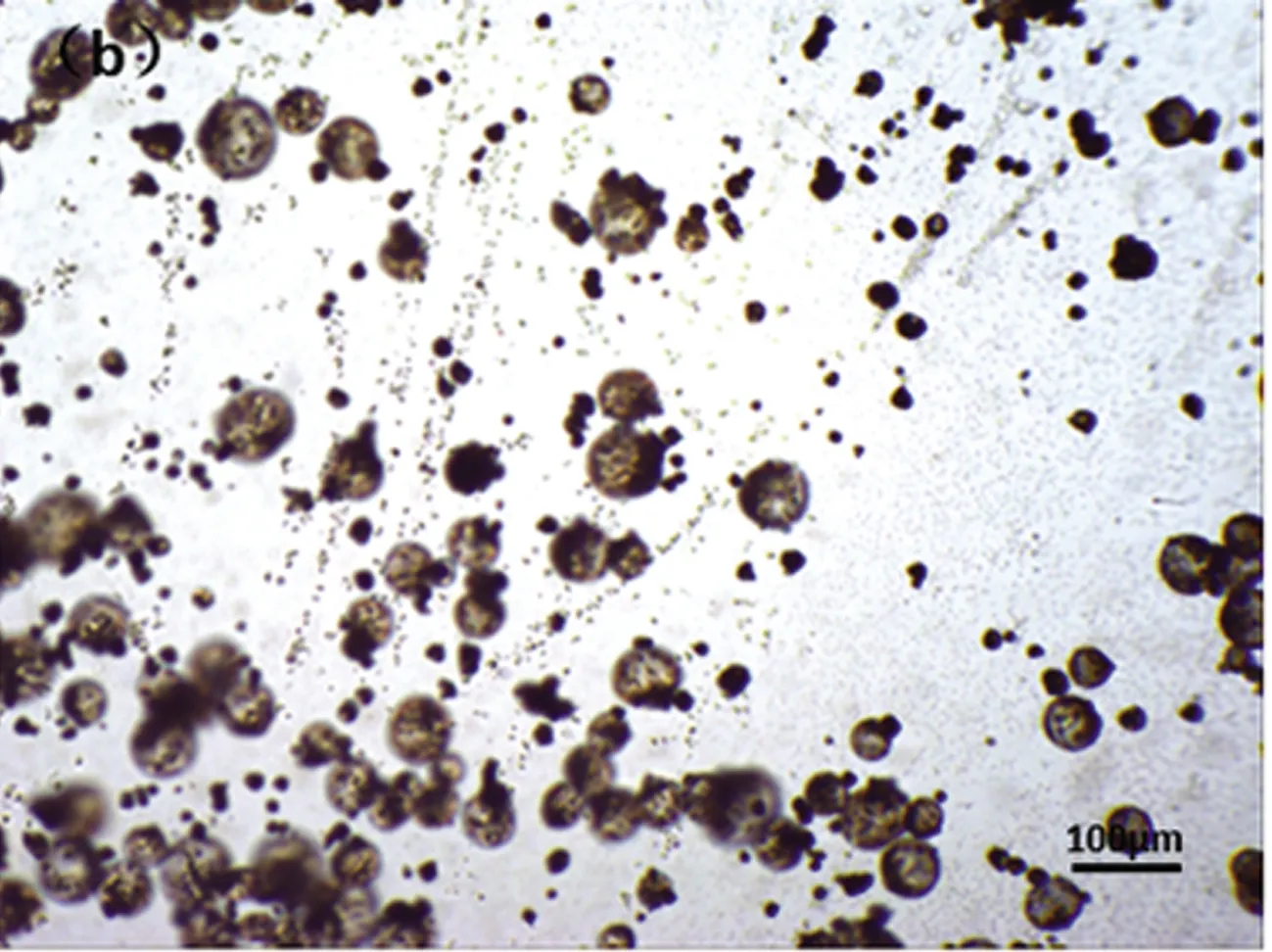
(b)开始发泡
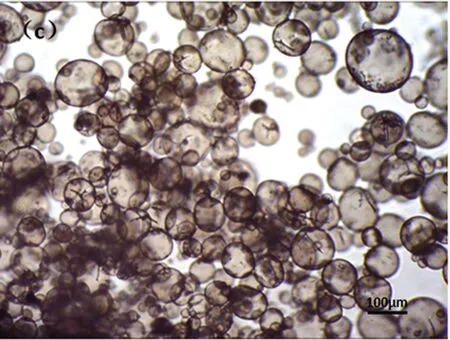
(c)最大发泡

(d)破泡图6 微球发泡过程Fig.6 Foaming process of microspheres
从图5可以看出:发泡微球粒径小、粒径分布窄,且其球形度好。
光学显微镜下,观察低温热膨胀微球发泡过程,从图6可以看出:微球发泡前后微球粒径相差大,发泡倍率高;微球开始发泡时间接近,微球大小相近,发泡均一。
3 结语
AB单体为4%时,膨胀微球具有最佳的发泡倍率、较好的稳泡温程和较好的粒径分布。异戊烷发泡剂为30%时,膨胀微球稳泡温程最佳,发泡倍率最佳,粒径分布较好。其起始发泡温度为98 ℃,终止发泡温度为136 ℃,稳泡温程为38 ℃,发泡倍率为75.7%。
