PD-L1胞外域密码子优化及原核表达纯化
2021-12-28王志华
方 宇 吕 舰 王志华
以T细胞为基础的免疫系统可识别和破坏异常细胞,如病原体感染的细胞和癌细胞。T细胞表面的T细胞受体(T cell receptor, TCR)与靶细胞表面的主要组织相容性复合(major histocompatibility complexes, MHC)的特异性结合在很大程度上受到一系列共刺激和共抑制受体及其配体(也称为免疫检查点)的控制[1]。免疫检查点通路通过调节抗原特异性T细胞的数量和功能活性,在限制组织损伤和维持自身耐受性方面发挥关键作用[2]。其中,程序性细胞死亡蛋白1(programmed cell death protein 1, PD-1)/程序死亡配体1(programmed death-ligand 1, PD-L1)通路因其被证明是大量恶性肿瘤的治疗靶点而脱颖而出[3]。
1992年,PD-1的基因结构被首次发现。大量研究发现,PD-1的特异性配体,PD-L1在许多恶性肿瘤细胞中大量表达,PD-1/PD-L1的结合会导致肿瘤细胞免疫逃逸[4]。T细胞在抗原刺激下活化,产生大量的细胞因子参与免疫反应。但同时产生的干扰素γ会导致肿瘤细胞表面PD-L1增多,肿瘤细胞表面的PD-L1与T淋巴细胞表面的PD-1发生特异性结合,会抑制效应T淋巴细胞的活性并诱导其发生凋亡,从而导致肿瘤细胞逃逸机体的免疫系统。因此,靶向PD-1/PD-L1免疫检查点,特异性阻断PD-1/PD-L1信号通路,可转化为临床抗肿瘤的方法。目前经FDA批准的PD-1/PD-L1单抗药物已用于治疗黑色素瘤、非小细胞肺癌、肾癌、霍奇金淋巴瘤、头颈部肿瘤、膀胱癌等[4~6]。
有研究报道,可通过指数富集的配基系统进化技术(systematic evolution of ligands by exponential enrichment, SELEX)筛选与靶蛋白特异性结合的DNA/RNA 适配体,通过阻断PD-1/PD-L1信号通路从而抑制肿瘤的免疫逃逸[7~10]。SELEX筛选的DNA/RNA 适配体存在以下优势:①可与靶蛋白实现特异性结合;②由于相对分子质量小,对肿瘤的穿透能力强,且在体内易降解消除,免疫原性较低;③与抗体比较,DNA/RNA适配体易于合成、运输、保存等。因此,针对PD-1/PD-L1的胞外结合区域筛选特异性的DNA/RNA 适配体,以阻断肿瘤的免疫逃逸,也为治疗恶行肿瘤提供了一种新思路。
本实验通过体外合成PD-L1胞外域对应的基因序列,重组转化至E.coli原核表达载体中,随后经IPTG诱导目的蛋白表达,通过密码子优化使目的蛋白在菌液上清中表达,最终采用镍离子螯合纯化树脂,得到了PD-L1胞外域目的蛋白,为后续筛选识别PD-L1胞外域特异性DNA/RNA 适配体提供实验基础。
材料与方法
1.质粒、菌株及细胞载体pET-28a(+),感受态E.coli DH5α和E.coli BL21(DE3)购自淼灵质粒平台;293T细胞取自武汉大学人民医院中心实验室。
2.主要试剂及仪器:DMEM培养基(高糖,含丙酮酸钠)、Gibco澳洲胎牛血清、蛋白marker、反转录试剂盒、超声破碎机购自赛默飞世尔科技(中国)有限公司;青霉素链霉素溶液购自武汉赛维尔生物科技有限公司;质粒DNA提取试剂盒、胶回收试剂盒购自天根生化科技(北京)有限公司;KOD-Plus-Neo购自东洋纺(上海)生物科技有限公司;限制性内切酶购自美国NEB公司;DNA marker、RNAiso Plus (trizol)试剂购自宝日医生物技术(北京)有限公司;琼脂糖购自广州赛国生物科技有限公司;卡那霉素、IPTG购自默克生命科学有限公司;镍离子螯合纯化树脂(complete His-Tag Purification Resin)购自上海罗氏制药有限公司。引物由生工生物工程(上海)股份有限公司合成。
3.目的基因扩增:293T细胞采用完全培养基(DMEM培养基加入10%的Gibco澳洲胎牛血清、1%的青霉素链霉素溶液)于37℃、5%CO2条件下培养。采用RNAiso Plus(trizol)试剂提取293T细胞总RNA,并根据试剂说明书将RNA反转录为cDNA,作为后续PCR反应的模板。根据NCBIGene子数据库中人源PD-L1(NM_014143)基因序列,采用Primer3Plus设计特异性引物。引物序列为:上游引物为5′-CCTGCAGGGCATTCCAGAAA-3′,下游引物为5′-TCCCTGCTTGAAGATCAGAAGT-3′。按KOD-Plus-Neo配制PCR反应体系,98℃预变性5min,98℃变性30s,60℃退火20s,68℃延伸30s,变性、退火、延伸35个循环,最后68℃再延伸5min,获取目的基因DNA片段。将PCR产物进行1.5%琼脂糖凝胶电泳,随后通用型DNA纯化回收试剂盒纯化目的片段。
4.重组质粒的构建:根据Uniport数据库中人源PD-L1的细胞膜外段(Position 19-238氨基酸),设计特异性重组引物。重组引物序列如下:上游引物为5′-TTAAGAAGGAGATATACCATGTTTACTGTCACG-GTTCCCAAG-3′,下游引物为5′-ATGGTCTTTGTAGTCGCTAGCCCTTTCATTTGGAGGATGTGC-3′。将上一步纯化得到的DNA片段作为模板,PCR反应体系同上,获取目的基因DNA片段。将PCR产物进行1.5%琼脂糖凝胶电泳,随后通用型DNA纯化回收试剂盒纯化重组目的片段。载体双酶切:将带有6×His标签的pET-28a(+)载体经XbaⅠ和XhoⅠ双酶切37℃水浴过夜,双酶切产物进行1 %琼脂糖凝胶电泳,随后通用型DNA纯化回收试剂盒纯化得到双酶切后载体。将重组目的片段与双酶切载体于37℃反应30min,进行重组反应。随后将重组产物转化至感受态E.coli DH5α,涂布于LB培养基(Kan+)上,于37℃培养箱培养12h。次日,将菌液送测序,将测序正确的菌液提取质粒DNA。最后将质粒转化入感受态E.coli BL21中,得到PD-L1表达菌株,命名为pET-28a-PD-L1-6×His,为后续原核表达做准备。
5.重组蛋白诱导表达条件筛选:将测序正确的pET-28a-PD-L1-6×His菌株置于37℃ 220r/min恒温摇床中过夜至平台期。次日按1∶50接种至5ml LB培养基(Kan+)中,37℃220r/min恒温摇床中培养至对数增长期,A600值为0.4~0.8,约1.5~2.0h,取少量诱导前菌液样本保留,随后按表1所示的诱导条件,继续诱导4h。取少量诱导后菌液样品保留,随后将剩余菌液12000r/min,离心3min,加入1ml 1×PBS重悬;冰浴超声,80%功率,超声2s,停2s,2~5min直至菌液清亮;再次离心12000r/min,离心5min,分离上清和沉淀,取少量上清及沉淀样品与之前保留的诱导前菌液、诱导后菌液一起与蛋白上样缓冲液混合均匀,95℃加热10min制备蛋白样本,进行Western blot法检测。
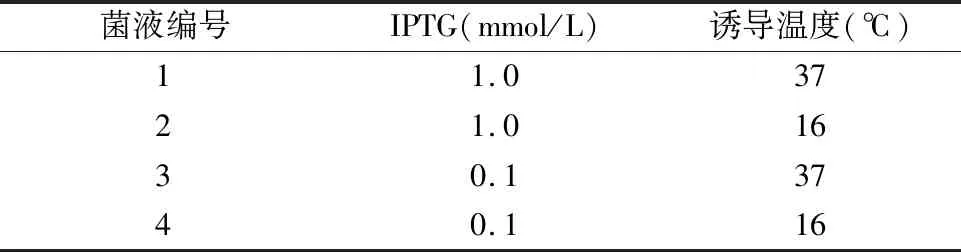
表1 诱导条件的筛选
6.大规模诱导目的蛋白表达:恒温摇床中过夜至平台期的pET-28a-PD-L1-6×His菌株按1∶50接种至500ml LB培养基(Kan+)中,37℃ 220r/min恒温摇床中培养至对数增长期,A600值为0.4~0.8,约1.5~2.0h。随后采用0.1mmol/L IPTG,37℃诱导温度诱导4h。将菌液分成两管,7500r/min离心3min收集菌液,分别加入20ml PBS重悬后,冰浴超声,80%功率,超声2s,停2s,1h直至菌液清亮, 离心收集上清或沉淀用于后续纯化过程。
7.包涵体洗脱及蛋白复性:若目的蛋白表达在沉淀中,便采用变性洗脱包涵体沉淀得到PD-L1目的蛋白。首先采用10ml Buffer A(50mmol/L Tris,pH值为8)重悬包涵体沉淀,12000r/min离心10min弃上清,清洗两遍;随后采用10ml Buffer B(50mmol/LTris、2mol/L尿素,pH值为8)重悬包涵体沉淀,12000r/min离心10min弃上清,清洗两遍;最后采用5ml变性液(0.1mol/L Tris、8mol/L尿素,pH值为8)重悬沉淀,37℃摇床孵育1h使包涵体完全溶解,12000r/min离心10min取上清,得到变性目的蛋白溶液。随后,将目的蛋白的变性溶液装于半透膜中,利用尿素浓度梯度配制复性液A~E,主要成分(1×PBS、10%甘油、2mmol/L还原型GSH、0.2mmol/L GS-SG氧化型)不变,尿素浓度由5、3、2、1、0mol/L依次递减,每遍均于4℃透析12h。
8.稀有密码子优化:为提高PD-L1胞外域在上清中诱导表达,进行天然蛋白的纯化,将PD-L1胞外域对应核苷酸序列进行稀有密码子优化。通用生物公司将稀有密码子替换、考虑GC比例,优化后的基因序列及氨基酸序列如下图(图1)。随后将优化后的目的基因序列构建至pET-28a表达载体中,转化至转化入感受态E.coli BL21中,得到NewPD-L1表达菌株,命名为pET-28a-newPD-L1-6×His。

图1 PD-L1基因序列及氨基酸序列优化前后对A.基因序列优化前后对比结果;B.氨基酸序列优化前后对比结果;1.优化前基因序列;2.优化后基因序列
9.镍离子螯合纯化树脂纯化目的蛋白:将大规模诱导后得到上清液进行后续的镍柱亲和层析纯化目的蛋白,具体纯化步骤如下。将3ml镍离子螯合纯化树脂用20ml平衡液(50mmol/L二水合硫酸二氢钠、300mmol/L氯化钠,pH值为8)进行柱平衡,流速1ml/min。随后,将上清液通过镍离子螯合纯化树脂,流速0.5ml/min,流通两遍。采用20ml流通液(50mmol/L二水合硫酸二氢钠、300mmol/L氯化钠、25mmol/L咪唑,pH值为8)通过树脂,流速1ml/min,去除树脂上非特异性蛋白。随后,用5ml洗脱液(50mmol/L二水合硫酸二氢钠、300mmol/L氯化钠、250mmol/L咪唑,pH值为8),流通树脂两遍,收集目的蛋白。最后,将目的蛋白洗脱液装入透析袋,于透析液中(1×PBS+5%甘油)4℃透析过夜,去除残留的咪唑,用蔗糖浓缩目的蛋白,液氮速冻后,-80℃保存。
结 果
1.PD-L1胞外域目的蛋白诱导条件筛选:本研究筛选了IPTG诱导浓度1mmol/L或0.1mmol/L,诱导温度37℃或16℃。实验结果表明,与诱导前的菌液比较,不同条件下诱导后的菌液在26kDa附近均有特异性目的蛋白过表达,表明目的蛋白能成功表达。此外,1mmol/L和0.1mmol/L IPTG均能诱导目的蛋白表达,两者比较差异无统计学意义。而在不同诱导温度下,PD-L1的蛋白表达比较差异有统计学意义,在37℃诱导温度下,目的蛋白的表达显著高于16℃(图2)。不同的诱导条件下,目的蛋白均在细菌裂解的沉淀中,而非裂解的上清中。因此,后续大规模诱导蛋白表达采用诱导温度37℃,0.1mmol/L IPTG。
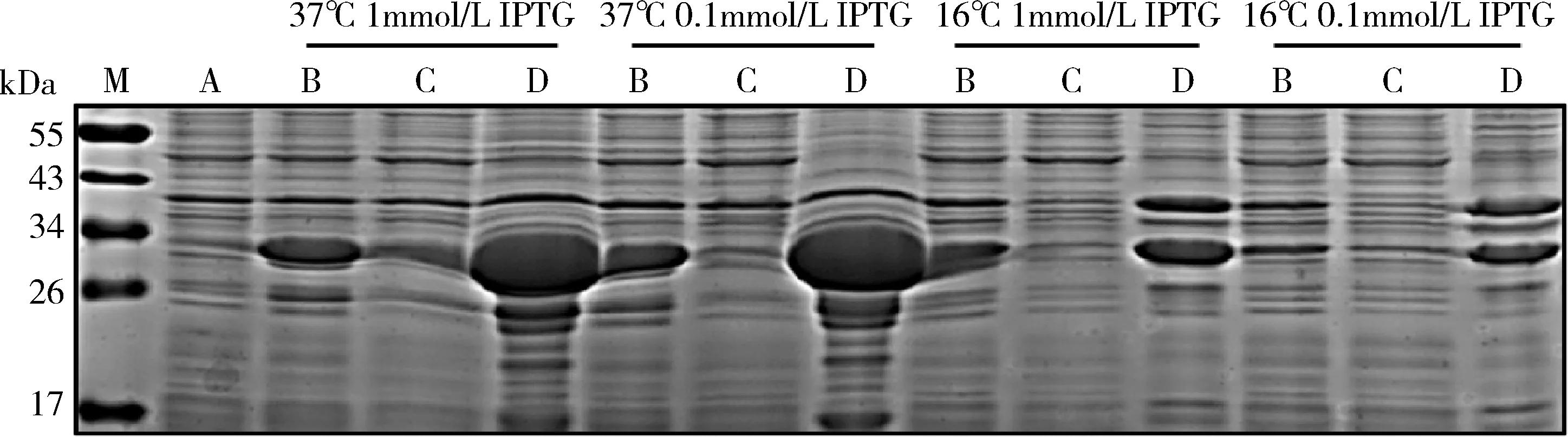
图2 PD-L1原核表达诱导条件筛选A.诱导前细菌样本;B.诱导后细菌样本;C.诱导后细菌裂解上清液;D.诱导后细菌裂解沉淀;M.蛋白marker
2.大规模诱导表达及包涵体变性纯化:于37℃,0.1mmol/L IPTG条件下大规模诱导表达,结果与图2一致,目的蛋白均能在沉淀中大量表达,因此后续采用溶解包涵体的方式纯化目的蛋白。结果如图3所示,对PD-L1胞外域目的蛋白的包涵体洗脱后,能得到纯度较高的目的蛋白,但在目的蛋白复性过程中,目的蛋白均析出。考虑到目的蛋白变性的洗脱方法无法成功纯化得到目的蛋白,为此需使目的蛋白表达于上清中,采用非变性的镍柱亲和层析纯化。因此,后续实验中对目的蛋白的基因序列进行了稀有密码子优化。
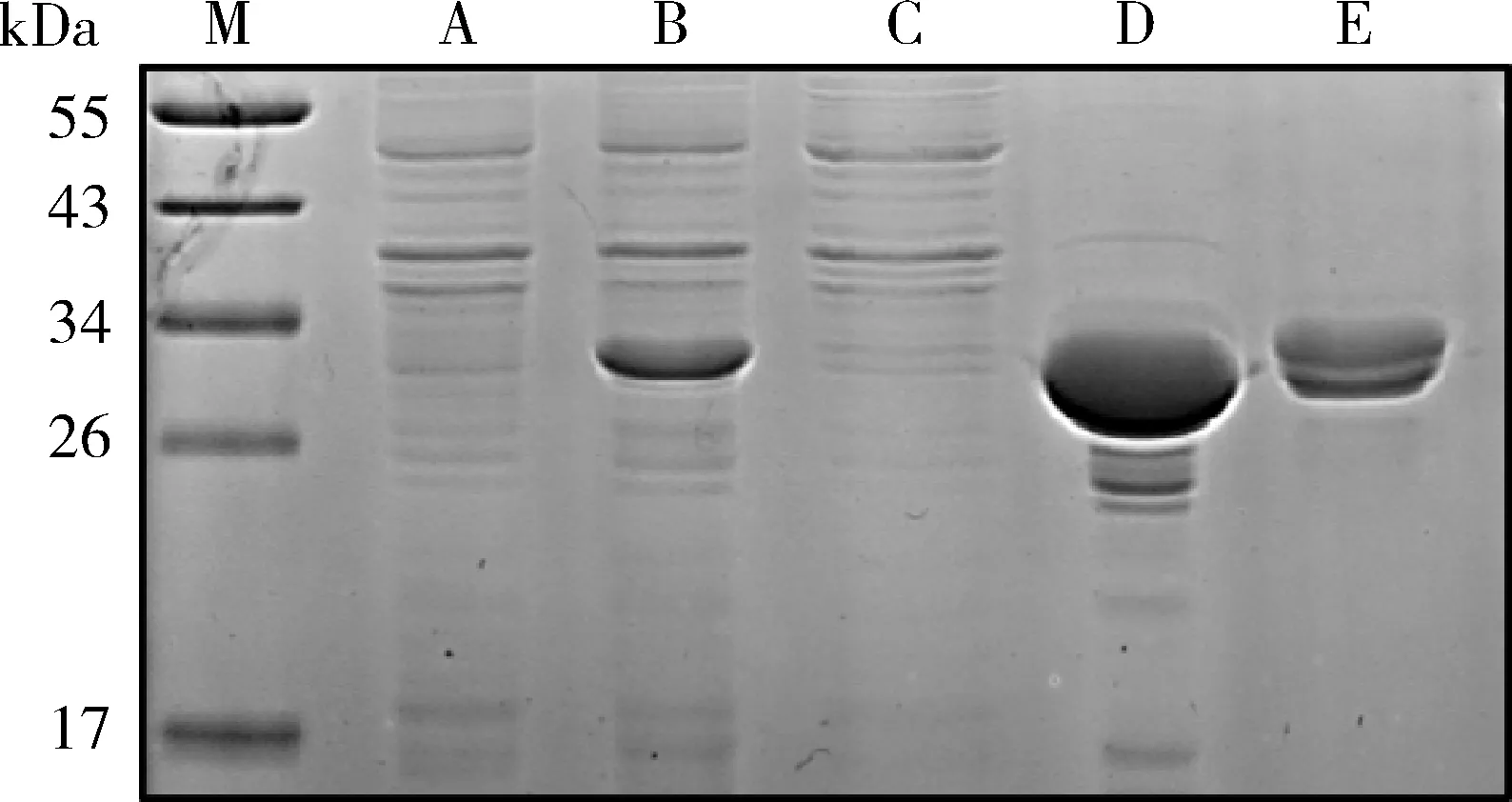
图3 PD-L1原核表达大规模诱导及包涵体洗脱A.诱导前细菌样本;B.诱导后细菌样本;C.诱导后细菌裂解上清液;D.诱导后细菌裂沉淀;E. 诱导后细胞包涵体洗脱;M.蛋白marker
3.密码子优化后目的蛋白诱导条件筛选:根据图1可以发现,利用密码子的简并性,稀有密码子的优化并未改变PD-L1胞外域蛋白氨基酸序列,优化后的PD-L1胞外域蛋白保持原有的抗原性及生物活性。经过稀有密码子优化后,笔者仍按表1方式筛选目的蛋白诱导表达条件。筛选结果如图4所示,1mmol/L和0.1mmol/LIPTG均能诱导目的蛋白表达,且两者比较差异无统计学意义。不同的诱导温度,目的蛋白的表达模式有显著差别。37℃的诱导温度下,目的蛋白在沉淀中大量表达,但在上清裂解液中无明显表达。16℃的诱导温度下,虽然目的蛋白的表达量大大减少,但在上清裂解液中表达成功。考虑到目的蛋白需采用非变性的镍离子螯合树脂纯化。因此,稀有密码子优化后采用的最佳诱导条件为16℃,0.1mmol/L IPTG,诱导12h。
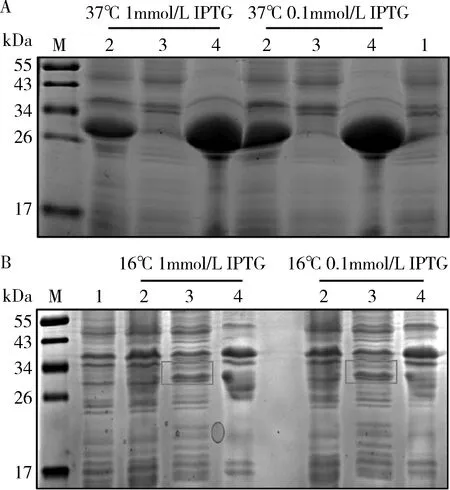
图4 基因优化后PD-L1原核表达诱导条件筛选A.诱导温度37℃的原核表达结果;B.诱导温度16℃的原核表达结果。1.诱导前细菌样本;2.诱导后细菌样本;3.诱导后细菌裂解上清液;4.诱导后细菌裂解沉淀;M.蛋白marker
4.镍离子螯合树脂纯化目的蛋白:经稀有密码子优化后,PD-L1胞外域目的蛋白于16℃,0.1mmol/L IPTG条件下进行大规模诱导表达,并将细菌超声裂解后的上清液通过镍离子螯合树脂进行亲和层析纯化。实验结果如图5,在该诱导条件下,目的蛋白仍大量表达于包涵体中,但有小部分以可溶方式表达于上清中,通过亲和层析后,成功得到了较高纯度的目的蛋白。综上所述,通过优化稀有密码子后,可使PD-L1胞外域目的蛋白少量表达于细菌裂解上清中,通过镍柱亲和层析可纯化纯度较高的目的蛋白,为后续的实验奠定了物质基础。
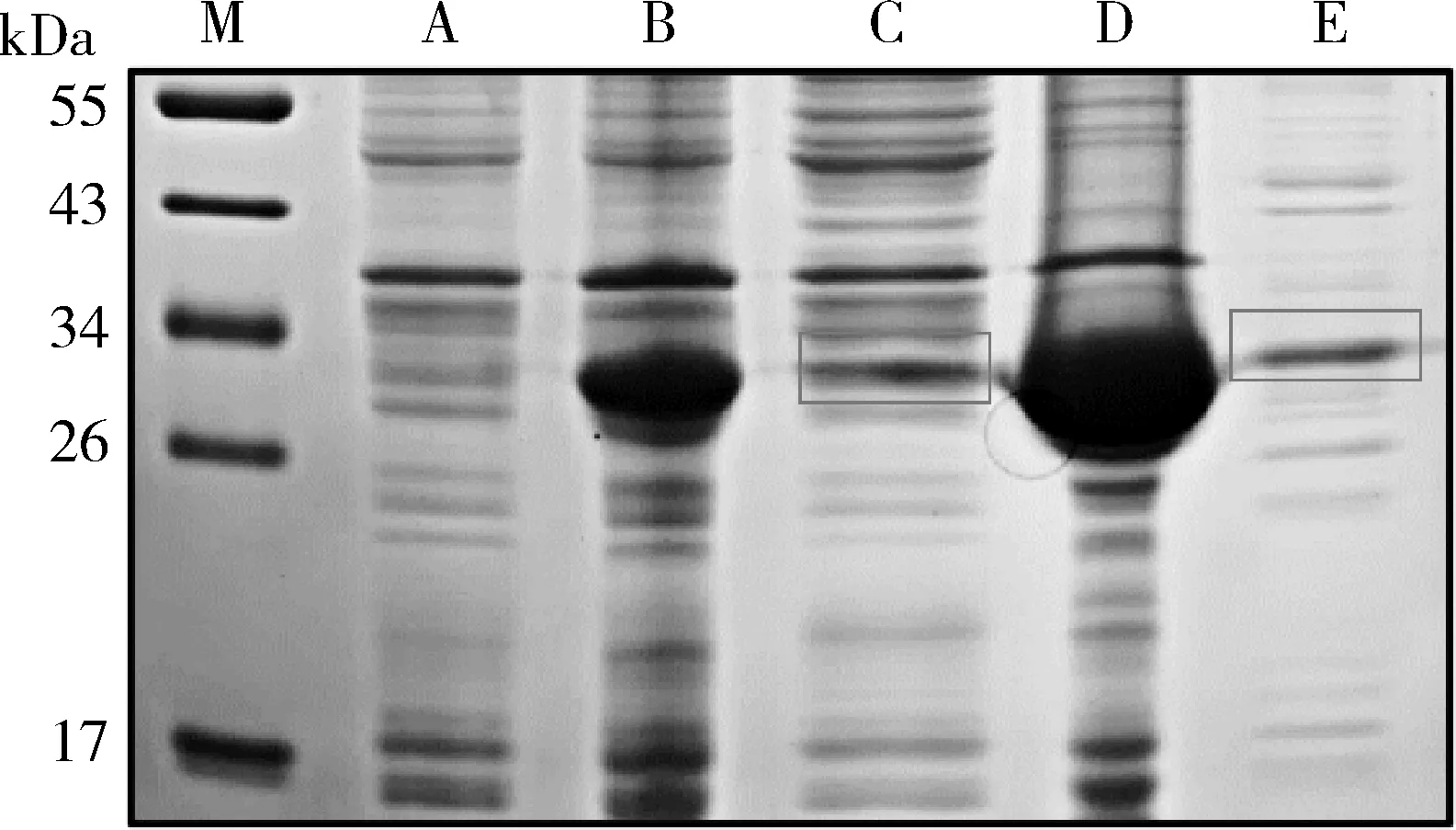
图5 PD-L1原核表达大规模诱导及亲和层析纯化A.诱导前细菌样本;B.诱导后细菌样本;C.诱导后细菌裂解上清液;D.诱导后细菌裂沉淀;E. 诱导后细菌裂解上清液亲和层析纯化;M.蛋白marker
讨 论
目前,PD-1/PD-L1免疫检查点疗法仍存在很多问题,如不同类型的肿瘤对PD-1/PD-L1抗体的敏感程度不同;免疫系统的非特异性激活[11~13]。某些患者接受治疗后加速肿瘤进程。SELEX技术可从随机单链核酸序列库中筛选出与靶蛋白特异性结合的核酸适配体,可为PD-1/PD-L1免疫检查点疗法提供一个新的思路[7~10]。本研究通过筛选IPTG诱导浓度、诱导温度、密码子优化,最终成功表达、纯化PD-L1胞外域片段,为后续的SELEX筛选PD-L1 适配体提供了实验基础。
与繁琐、复杂且蛋白得率低的真核表达系统比较,基于大肠杆菌的原核表达系统简单易行、目的蛋白得率高,已成为获取目的蛋白的常规方法[14~17]。本研究发现,在密码子优化前后,0.1mmol/L与1mmol/L IPTG诱导浓度均可成功诱导目的蛋白表达,且两者间无明显差异。而诱导温度不仅影响目的蛋白的诱导效率,还会影响目的蛋白的表达模式。笔者发现在密码子诱导前后,诱导温度越高,目的蛋白的诱导效率越高。此外,通过稀有密码子优化后,16℃低温诱导表达可使目的蛋白表达于细菌裂解上清中,可能是因为低温使蛋白表达效率降低的同时,改善了蛋白的空间构想,使其正常折叠。
真核生物的蛋白在大肠杆菌原核表达系统中,由于缺乏某些蛋白质折叠辅助因子或环境不适,易形成包涵体[11]。此外,不同物种间存在明显的密码子偏好,若使用频率低的稀有密码子大量存在,会严重减小目的蛋白的表达水平,甚至会造成点突变或移码突变表达出序列错误的多肽[12, 18~20]。目的蛋白中含有大量稀有密码子,最直接、高效的方法是对目的蛋白进行稀有密码子优化,可改善蛋白的空间构象,使其正常折叠,从而减少包涵体的形成并保持生物活性实现可溶性的表达[21]。本研究发现在密码子优化前,在最优筛选条件下,PD-L1蛋白仍在包涵体中表达,并在蛋白复性析出最终无法得到目的蛋白。而在密码子优化后,采用16℃,0.1mmol/L IPTG诱导12h可使PD-L1目的蛋白以可溶状态少量表达于细菌裂解上清液中,随后通过镍离子螯合纯化树脂得到了少量的PD-L1胞外域目的蛋白。
综上所述,本研究表达、纯化了PD-L1胞外域目的片段,为PD-L1的其他相关研究提供了物质基础,为后续筛选PD-L1胞外域特异性结合的核酸适配体提供了实验基础。SELEX利用该技术可以从随机单链核酸序列库中筛选出特异性与靶物质高度亲和的核酸适体[7~10]。若利用SELEX技术筛选得到的特异性适配体能阻断PD-1/PD-L1间的结合,则能为肿瘤逃逸的防治提供新的思路与方向。
