犬博卡病毒结构蛋白VP1多克隆抗体的制备及特异性鉴定
2021-12-27孙锦涵张淋然丁彦琴孙玉宁
穆 乐,孙锦涵,张 健,张淋然,丁彦琴,孙玉宁
(1.宁夏医科大学基础医学院,银川 750004;2.海南医学院临床医学院,海口 571199)
犬博卡病毒(Minute virus of canine,MVC)属于细小病毒家族的博卡病毒亚家族成员之一。博卡病毒属主要含有牛博卡病毒(Bovine parvovirus,BPV)、犬博卡病毒(MVC)以及人博卡病毒(Human bocavirus,HBoV)3个家族成员。近年来,又相继在黑猩猩(Gorilla bocavirus,GBoV)、猪(Porcine bocavirus,PBoV)、猫(Feline bocavirus,FBoV)、犬(Canine bocaviruses,CBoV)以及加利福尼亚海狮中发现了博卡病毒的新家族成员[1-4]。细小病毒是一种无包膜、分子量小、线性单链的DNA病毒,基因组大小为5 kb[5]。MVC基因组全长为5402 nt,包含3个开放阅读框(open reading frame,ORF),分别编码非结构蛋白NS1(85 kDa)、NP1(20 kDa),结构蛋白VP1(81 kDa)、VP2(63 kDa)、VP3(61 kDa)[6]。
VP1和VP2蛋白是细小病毒衣壳的主要结构蛋白,其中VP2蛋白约占病毒粒子衣壳蛋白的90%,VP1蛋白约占10%[7]。细小病毒的VP1和VP2基因属于重复序列结构基因,它们编码的结构蛋白存在一段相同的氨基酸序列:VP1蛋白在N端多出一段氨基酸序列,是VP1蛋白所独有序列(独特区),其后续的氨基酸序列与VP2氨基酸序列一致。研究发现,细小病毒VP1和VP2蛋白参与到病毒感染宿主细胞的过程,并且VP1蛋白在病毒衣壳蛋白的合成及形成感染性颗粒的过程中起着重要作用。VP1蛋白N端的独特区域含有磷脂酶A2(PLA2)的保守序列(如HDXXY、YXGXG等区域),具有磷脂酶A2活性,其功能对于病毒DNA从吞噬体进入细胞核发挥重要作用[4,8]。博卡病毒 VP1在N端前有132个氨基酸是VPl蛋白所独有的,剩余571个氨基酸与VP2氨基酸序列完全一致。本研究前期通过突变的方法,改变MVC的磷脂酶A2的保守序列氨基酸,发现酶活性的改变明显影响了MVC在宿主细胞中的DNA复制能力[6],说明VP1结构蛋白在MVC基因组复制中发挥作用。本实验通过构建MVC结构蛋白VP1 N端独特区序列的原核表达载体,经诱导表达并获得纯化的融合目的蛋白,免疫新西兰大白兔后制备VP1的多克隆抗体,为后续开展MVC在感染及致病机制等方面的研究提供物质基础。
1 材料与方法
1.1 材料大肠杆菌DH5α和BL21(DE3)、质粒pET32a(+)均由宁夏医科大学基础医学院生化实验室保存;沃尔特里德犬细胞(walter reed dog,WRD)及博卡病毒由美国堪萨斯大学医学院微生物系邱建明教授惠赠;新西兰大白兔购自宁夏医科大学动物实验中心。
1.2 原核表达载体构建应用BioEdit软件设计引物,引物由生工生物工程(上海)股份有限公司合成。上游引物:5'-GGTCTCGAATTCATGGCTCCTCC GAATAGAAAACC-3'(下划线标记为EcoRⅠ酶切位点),下游引物:5'-GGTCTCCTCGAGCTACC TAGCTTGTTTAGAG-3'(下划线标记为XhoⅠ酶切位点),以前期获得的感染性克隆载体pI-MVC为模板扩增VP1 5'端独特区基因片段(396 nt)。扩增条件:95℃变性30 s,60℃退火30 s,72℃延伸35 s,共30个循环;最后72℃延伸10 min。回收并纯化PCR片段,将纯化产物和pET32a(+)优化载体经EcoRⅠ/XhoⅠ双酶切并胶回收后连接转化至大肠杆菌DH5α中。挑选阳性单克隆菌落,提取质粒,双酶切鉴定,并送至生工生物工程(上海)股份有限公司进行测序。
1.3 重组蛋白的表达及纯化采用电转化法将构建的原核表达重组质粒pET32a-VP1N转化至BL21感受态细胞中,将菌液涂布LB固体培养基后置于37℃温箱孵育;次日,挑取阳性单克隆至LB培养液(氨苄青霉素抗性)中培养;然后按1∶100扩增培养至吸光度值为0.7~0.8。取1 mL培养物作为未诱导阴性对照组。经终浓度为0.25 mmol/L的IPTG诱导,37℃震荡培养4 h。于4℃条件下10 000×g离心收集诱导表达培养物。通过10%SDSPAGE电泳分析目的蛋白的表达情况,并用镍柱纯化融合蛋白。蛋白定量测定后于-80℃冰箱中保存备用。
1.4 免疫动物选取2.0 kg左右健康新西兰大白兔,取500 μg的纯化融合蛋白用PBS稀释后与等体积的弗氏完全佐剂充分混合,于家兔足部、背部皮下多点注射。2周后再以相同剂量的蛋白与弗氏不完全佐剂充分混合,进行加强免疫;4周后,每隔7 d取250 μg纯化蛋白加强免疫1次,随后处死兔子取全血,分离抗血清,进行Protein A和去交叉的His柱纯化。
1.5 抗血清纯化从冰箱取出Protein A柱和去交叉的His柱,室温放置几分钟后用5倍柱体积的二级水过柱及相同体积的20 mmol/L的磷酸钠溶液(pH7.0)平衡柱子。离心好的血清过Protein A柱,经10倍柱体积的20 mmol/L的磷酸钠溶液(pH7.0)洗柱后,用4倍柱体积100 mmol/L的甘氨酸(pH2.5)洗脱柱子,收集洗脱液并用0.2倍柱体积1 mol/L Tris-HCl(pH9.0)充分混匀。然后洗脱液(约8.4 mL)过His交叉柱,收集洗脱液,此为纯化后目的抗体溶液。加入等体积的甘油后分装,于-80℃冰箱保存备用。
1.6 ELISA法测定多抗效价已纯化的蛋白抗原包被浓度1 μg/mL,多肽抗原包被浓度 4 μg/mL。1%BSA封闭。将纯化的多克隆抗体血清分别按1∶3125、1∶6250、1∶12 500、1∶25 000、1∶50 000、1∶100 000、1∶200 000、1∶400 000、1∶800 000、1∶1 600 000的倍数稀释后,一抗37℃孵育1 h,二抗37℃孵育45 min。加显色液反应15 min,终止反应后测定450 nm处的吸光度值。
1.7 Western blot检测VP1N抗体的特异性WRD细胞感染MVC病毒48 h后,提取蛋白后进行SDS-PAGE电泳,湿转法转移至PVDF膜上。将PVDF膜于5%脱脂奶粉封闭,室温封闭1 h;PBST洗涤3次,每次5 min;分别加入制备的VP1抗体(VP1N,1∶2000)和VP2抗体(即VP1C,1∶2000,实验室先期制备的抗体),4℃孵育过夜;PBST洗涤3次,每次5 min;然后将PVDF膜转移到山羊抗兔的二抗孵育液(1∶400)中,在室温条件下孵育1 h,PBST洗涤3次,每次5 min。用ECL化学发光法进行检测。
1.8 免疫荧光法鉴定VP1N抗体特异性用6孔板(含有盖玻片)进行WRD细胞培养及爬片,待细胞贴壁后加入MVC感染,同时设置MVC未感染组(阴性对照组)。感染48 h后,吸掉培养液并用PBS清洗细胞3次,加入冰冷的细胞固定液(丙酮和甲醇1∶1混合),室温固定10 min。随后PBS洗涤3次,每次10 min;加入0.5%的TritonX-100作用10 min;PBS洗涤3次,每次10 min;山羊血清于37℃封闭20 min;以纯化的兔抗VP1多克隆抗体作为一抗(1∶50),37℃孵育2 h;PBS洗涤3次,每次10 min;加FITC标记的山羊抗兔IgG(1∶50),室温避光孵育1 h;DAPI染色10 min,封片后在荧光显微镜下观察结果。
2 结果
2.1 重组质粒pET-32a ( + ) -VP1N的酶切鉴定重组质粒pET-32a(+)-VP1N经EcoRⅠ和XhoⅠ双酶切后,产物经1%琼脂糖凝胶电泳,在400 bp大小位置可见清晰的条带,表明VP1N基因片段成功插入到pET32a(+)载体中,同时经测序鉴定,证实重组质粒pET-32a(+)-VP1N构建成功(图1)。

图1 重组质粒pET-32a (+) -VP1N的酶切鉴定Fig.1 Identification of recombinant plasmid pET-32a(+)-VP1N by enzyme
2.2 目的蛋白的诱导表达及纯化重组表达质粒pET-32a(+)-VP1N在大肠杆菌中经I PTG诱导后,经SDS-PAGE电泳在34 kDa处出现一条明显的蛋白条带,大小与融合蛋白的理论值一致,而未诱导的重组质粒转化菌在相应的位置未出现相同的条带。扩大培养并进行诱导后,超声破碎后离心分离沉淀和上清液,SDS-PAGE电泳可知重组蛋白主要以可溶性的形式存在于上清液中。利用Ni-NTA亲和层析柱纯化融合蛋白,得到单一条带(图2)。

图2 SDS-PAGE检测VP1N重组蛋白的表达和纯化Fig.2 Expression and purification of fusion protein of VP1N detected by SDS-PAGE
2.3 VP1N抗血清效价测定以免疫前新西兰大白兔血样标本的血清作为对照。用ELISA法测定多克隆抗体效价,结果表明,VP1N抗体效价可达1∶1 600 000(表1)。

表1 ELISA法检测VP1N蛋白抗血清效价Table 1 Titers of anti-VP1N serum detected by ELISA
2.4 Western blot法检测VP1N多克隆抗体特异性病毒VP1基因位于MVC基因组的第3081~5192 nt,VP2基因位于MVC基因组的第3477~5192 nt(图3A)。因此,VP1和VP2基因属于含有相同序列的基因,VP1较VP2基因在病毒基因组5'端长396 nt,编码132个氨基酸;除此外,VP1和VP2的其他基因序列及氨基酸序列完全一致,编码571氨基酸。本次研究扩增VP1基因5'端独特区的396 nt核苷酸序列,并成功构建pET-32a(+)-VP1N重组载体,免疫新西兰大白兔后得到的VP1抗体(即VP1N抗体)仅可特异性识别病毒VP1蛋白,不识别VP2蛋白。前期我们以VP1/VP2蛋白的C端构建重组体并制备了VP2抗体(即VP1C抗体),此抗体能够同时识别病毒的VP1和VP2蛋白(图3A)。通过Western blot检测VP1 N端抗体和VP1 C端抗体的特异性,结果显示,MVC感染WRD细胞后,VP1C抗体检测到两条特异性条带,大小分别为81 kDa和63 kDa,与病毒VP1和VP2结构蛋白大小一致;VP1N抗体只在81 kDa处可以清晰地看到一条特异性条带,与VP1蛋白大小一致,VP1C抗体可以同时识别VP1和VP2两种结构蛋白。研究结果表明,此次制备的VP1N抗体仅识别VP1蛋白,具有非常高的特异性。
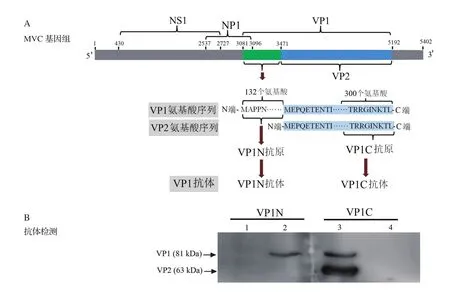
图3 VP1N抗体靶向序列及Western blot特异性分析Fig.3 VP1 antibody targeting sequence and specificity analysis of VP1N polyclonal antibodies
2.5 免疫荧光法检测VP1N抗体特异性MVC感染WRD细胞24 h后,利用PBS缓冲液清洗细胞,固定后采用免疫荧光技术分析VP1N抗体活性。结果显示,在MVC感染组中,出现特异性的绿色荧光(主要定位于细胞核中),未感染组中未见绿色荧光。结果说明,VP1N多克隆抗体能够与VP1蛋白发生特异性反应,进一步说明VP1N多克隆抗体具有较高的特异性(图4)。

图4 免疫荧光法检测VP1N多克隆抗体的特异性Fig. 4 Specificities of VP1N ployclonal antibodies analyzed by immunofluorescence assay
3 讨论
博卡病毒为最早于1967年从一只健康的德国牧羊犬的粪便样本中发现并分离得到的一种细小病毒[9]。此后,在亚洲国家的韩国、日本以及中国上海发现MVC感染的病例报道[10]。目前,关于MVC感染分布与犬类疾病的相关性尚未完全阐明。早期的研究发现,MVC作为新生幼犬和犬胚胎期感染疾病的重要致病原,感染后可导致流产、出生畸形,甚至胚胎死亡和再吸收[11-13]。同时也有研究者认为,MVC感染引起疾病可能与产生免疫抑制有关,发现患病犬出现包括单核细胞吞噬功能明显减少、病毒血症、肺泡细胞肥大和增生的间质性肺炎、水肿、坏死和炎症等症状[14-16]。近来也有研究发现,MVC感染与犬的神经性疾病和严重的胃肠炎疾病有关[17-18]。随着人民生活水平的提高,越来越多的家庭开始养宠物,尤其以家犬较多。因此,针对家犬中博卡病毒感染的检测、治疗值得大家关注。
在前期研究中,本课题组成功获得了MVC基因组包含的两个末端发夹回文结构在内的全部碱基序列(5402 nt),并首次成功构建了博卡病毒MVC感染性克隆载体pI-MVC,分析了MVC编码的非结构蛋白NS1、NS2和NP1基因在基因组中的基本结构特点、转录特性及基本功能[6]。结构蛋白VP1位于MVC基因组核酸序列的第3081~5192 nt位,含有2112个碱基,翻译编码704个氨基酸,蛋白质的分子量约为81 kDa;结构蛋白VP2位于MVC基因组的第3477~5192 nt位,含有1716个碱基,翻译编码571个氨基酸,蛋白质的分子量约为63 kDa。MVC病毒的VP1和VP2基因序列也属于重复基因序列,VP1基因较VP2基因在5'端长396 nt,多编码132个氨基酸,其他基因序列和氨基酸序列与VP2完全一致,编码571氨基酸。我们前期研究中,针对VP1近C端的300氨基酸序列制备了多克隆抗体[19],由于VP1和VP2蛋白C端氨基酸序列是完全一样,靶向VP1C的抗体能够同时特异性的识别VP1和VP2两种蛋白质,因此,我们在前期研究中针对VP1近C端的300个氨基酸序列制备了多克隆抗体,可以同时识别VP1和VP2蛋白(图3)。本次研究中,我们针对VP1蛋白N端特异的132个氨基酸序列进行克隆,并成功构建原核表达载体,通过诱导表达、纯化融合蛋白,免疫新西兰大白兔后得到可特异性识别VPI蛋白的高滴度多克隆抗体VP1N,效价达到1∶1 600 000。同时,通过Western blot检测抗体的反应性和特异性可知,VP1N抗体仅能识别VP1蛋白,而与VP2蛋白没有反应,由此说明制备的VP1N抗体具有非常好的抗体活性和特异性。同时,应用免疫荧光检测发现,VP1N抗体同样能够特异性检测到MVC感染的WRD细胞中VP1 蛋白的表达。
综上所述,本研究成功构建犬博卡病毒结构蛋白VP1独特区的原核表达载体,制备的VP1N多克隆抗体具有非常好的抗体活性和特异性。该研究结果为进一步加深对博卡病毒感染、致病机制及VP1基因功能等的研究提供了实验基础和理论支持。
