Effects of immune cells on mesenchymal stem cells during fracture healing
2021-12-24SabrinaEhnertBornaReljaKatharinaSchmidtBleekVerenaFischerAnitaIgnatiusCarenLinnemannHelenRinderknechtMarkusHuberLangMiriamKalbitzTinaHistingAndreasNussler
Sabrina Ehnert, Borna Relja, Katharina Schmidt-Bleek, Verena Fischer, Anita Ignatius, Caren Linnemann,Helen Rinderknecht, Markus Huber-Lang, Miriam Kalbitz, Tina Histing, Andreas K Nussler
Sabrina Ehnert, Caren Linnemann, Helen Rinderknecht, Tina Histing, Andreas K Nussler, Siegfried Weller Research Institute at the BG Trauma Center Tübingen, Department of Trauma and Reconstructive Surgery, University of Tübingen, Tübingen 72076, Germany
Borna Relja, Experimental Radiology, Department of Radiology and Nuclear Medicine, Ottovon-Guericke University, Magdeburg 39120, Germany
Katharina Schmidt-Bleek, Julius Wolff Institute and Berlin Institute of Health Center of Regenerative Therapies, Charité - University Medicine Berlin, Berlin 13353, Germany
Verena Fischer, Anita Ignatius, Institute of Orthopedic Research and Biomechanics, Ulm University Medical Center, Ulm 89091, Germany
Markus Huber-Lang, Institute for Clinical and Experimental Trauma-Immunology (ITI), University Hospital Ulm, Ulm 89091, Germany
Miriam Kalbitz, Department of Trauma and Orthopedic Surgery, University Hospital Erlangen Friedrich-Alexander University Erlangen-Nürnberg (FAU), Erlangen 91054, Germany
Abstract In vertebrates, bone is considered an osteoimmune system which encompasses functions of a locomotive organ, a mineral reservoir, a hormonal organ, a stem cell pool and a cradle for immune cells. This osteoimmune system is based on cooperatively acting bone and immune cells, cohabitating within the bone marrow. They are highly interdependent, a fact that is confounded by shared progenitors, mediators, and signaling pathways. Successful fracture healing requires the participation of all the precursors, immune and bone cells found in the osteoimmune system. Recent evidence demonstrated that changes of the immune cell composition and function may negatively influence bone healing. In this review, first the interplay between different immune cell types and osteoprogenitor cells will be elaborated more closely. The separate paragraphs focus on the specific cell types, starting with the cells of the innate immune response followed by cells of the adaptive immune response, and the complement system as mediator between them. Finally, a brief overview on the challenges of preclinical testing of immunebased therapeutic strategies to support fracture healing will be given.
Key Words: Trauma; Bones; Immune response; Mesenchymal stem cells; Fracture healing
INTRODUCTION
In vertebrates, bone can be considered as an osteoimmune system encompassing functions of a locomotive organ, a mineral reservoir, a hormonal organ, a stem cell reservoir and a cradle for immune cells. This osteoimmune system is based on cooperatively acting bone and immune cells, cohabitating within the bone marrow. They are highly interdependent, a fact that is confounded by shared progenitors and signaling pathways. Receptor activator of nuclear factor kappa B ligand (RANKL) is a well described example of this interdependency. Well known as a key factor for osteoclast differentiation, RANKL also regulates T-cell differentiation and proliferation[1]. Mesenchymal stem cells (MSC) are adult stem cells. In bone, MSCs are traditionally found in the bone marrow. They act as precursor cells for chondrogenic, adipogenic and osteogenic cells during bone homeostasis and fracture healing[2]. However, MSCs also play crucial roles in hematopoiesis and maintenance of immune cell progenitors[3]. Recently, different MSC subsets with distinct roles on different immune cell progenitors have been described[4,5]. In MSCs, this process seems to be strongly dependent on the expression of stem cell growth factor, also known as C-type lectin domain family 11 member A or osteolectin. MSCs lacking this factor seem to regulate hematopoiesis, while MSCs expressing osteolectin strongly influence maintenance and function of common lymphoid progenitor cells and their progenies[4]. Furthermore, these osteolectin positive MSCs, prone to undergoing osteogenesis[5], increase in number following a bone fracture and thus, represent the main source of osteoprogenitor cells during fracture healing[4].
The bone-immune cell interplay at first considered the role of the innate immune system but more and more the role of the adaptive immune system was investigated. With respect to the adaptive immune system, the question arises whether it has beneficial or detrimental effects on regeneration. Considering evolution, the development of the immune system coincides with a decline in the regenerative capacity —i.e.the more elaborate the immune response the less capable of regeneration an organism is[6]. A hallmark of adaptive immunity is its capacity to “remember” pathogens. This immune memory is only present in vertebrates, which constitute 1%-2% of the living species[7]. Indeed, while their immune system is very elaborate, vertebrates only have a very limited regenerative capacity. An exception are mammalian embryos, which, still lacking adaptive immune responses, are capable of scar-free regeneration, a capacity that diminishes after birth and with aging[8]. This indicates that adaptive immunity is rather unfavorable for regeneration.
In human adults, most tissues heal with scar tissue formation. Bone, however, is capable of healing without scar tissue formation. Bone healing is a highly complex process consisting of numerous well-orchestrated interdepending and overlapping steps, which, if undisturbed, result in tissue fully restored in form and function[9]. Using endochondral bone regeneration as a model, effects of the innate and adaptive immune system have been investigated to understand their role in regenerative processes and to explore the possibility of using immune-modulatory strategies for the development of new therapeutic approaches. Upon bone injury and inadvertent vessel rupture, the initial step in healing consists of coagulation and the coupled release of platelet-derived pro-inflammatory cytokines,e.g., interleukin (IL)-6, or tumor necrosis factor (TNF)-α[10]. The main functions of the clotting reaction are the closure of a possible breach of the outer hull of an individual and a strong defense against possible invading pathogens. The released cytokines stimulate homing of leukocytes into the fracture site (Figure 1). With vessel rupture, the supply of oxygen and nutrients is diminished at the injury site. Thus, cells active in this initial phase of bone healing need to be capable of functioning in this detrimental environment (low pH, low oxygen, disturbed sodium and potassium balance[11,12]).
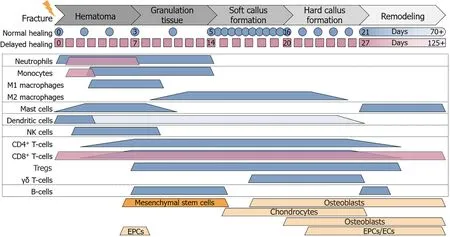
Figure 1 Cell composition at the site of fracture.
Neutrophils, the most abundant immune cells, are the first cells to arrive at inflammatory sites. The fracture hematoma was shown to be a strong inducer of neutrophil homing[13,14]. Within less than an hour, they are recruited to the site of fracture[15], triggered by damage associated molecular patterns (DAMPs), including mitochondrial DNA (deoxyribonucleic acid) fragments[16]. Equipped with highly potent activities such as phagocytosis, respiratory burst, or neutrophil extracellular trap (NET) formation, neutrophils strongly contribute to the first inflammatory reaction and formation of granulation tissue. Additional recruitment of fresh neutrophils from bone marrow by the C-X-C chemokine receptor (CXCR) type 4 (CXCR4)-C-X-C motif chemokine ligand (CXCL) 12 (CXCL12) axis, further supports the inflammatory reaction[17]. This inflammatory reaction at the beginning of the healing process has a substantial influence on the whole process[18]. As a consequence of the systemic inflammatory reaction, neutrophils invade not only the site of injury but also lung and liver tissue rapidly after a fracture[19]. Almost simultaneous to neutrophils, mast cells (MCs) and dendritic cells (DCs) appear in the fracture hematoma. As tissue-resident hematopoietic cells, MCs modulate innate and adaptive immune responses, not only in the early inflammatory phase of fracture healing, but also later during bone remodeling, where MCs exert several functions in the regulation of angiogenesis, osteogenesis, and osteoclastogenesis[20]. Shortly after, natural killer (NK) cells arrive at the site of injury, attracted by pro-inflammatory cytokines TNF-α and IL-6[21]. The role of NK cells in fracture healing is not fully understood and seems strongly dependent on the current inflammatory status. Within one day, monocytes are recruited to the fracture hematoma. They first differentiate into pro-inflammatory M1 macrophages, contributing to the clearance of the granulation tissue by phagocytosis. During callus formation, anti-inflammatory M2 macrophages are present, supporting migration of osteoprogenitor cells, matrix formation, and angiogenesis[22]. As precursors for osteoclasts, monocytes/macrophages also contribute to the final remodeling phase of fracture healing. Osteoclast differentiation is strongly dependent on factors secreted by B- and T-lymphocytes, which invade the fracture site, when granulation tissue is fully accomplished. In their pro-inflammatory state, T cells may act in the fracture environment[23,24].
This initial pro-inflammatory reaction is required to initiate fracture healing, not only for defense purposes but also to attract cells needed in the downstream healing process. However, this process needs to be tightly monitored, because the pro-inflammatory reaction becomes detrimental if it is too strong or lasts too long[25]. Under anaerobic conditions the transcription factor, hypoxia inducible factor 1α, is stabilized and induces the expression of angiogenic factors by cells in the fracture site. This initiates the essential revascularization step by homing of endothelial progenitor cells (EPCs). Expression analysis revealed that upregulation of angiogenic factors is paralleled by enhanced anti-inflammatory signaling, to terminate the initial proinflammatory reaction in order to proceed towards the next healing phase[9]. A switch from pro- to anti-inflammatory signaling is achieved,i.e., by an upregulation of IL-4 and IL-13, which support the Th2 (T helper cells type 2) and M2 phenotype in T cells and macrophages[26]. Influencing the initial immune reaction by local application of these cytokines in a mouse osteotomy model significantly improved healing[22], emphasizing the importance of a tightly regulated initial inflammatory reaction and of swift downregulation of the initial pro-inflammatory reaction for a successful healing outcome. Thus, repeated irritation of the fracture hematoma can favor non-union[27]. Furthermore, movement of the early fracture hematoma into muscle tissue can induce ectopic bone formation[28], underlining the importance of the hematoma in the bone forming process.
The initial pro-inflammatory phase is required to attract progenitor cells,e.g., EPCs or MSCs, to the site of fracture. MSCs with their manifold immunomodulatory and regenerative properties serve as progenitors for fibroblasts, chondrocytes and osteoblasts involved in the following callus formation. NK-, B-, and T-cells contribute to the effect of MSCs during this phase of fracture healing. Osteoblasts are the bone forming cells, producing collagen I that serves as the matrix for mineralization. In a mouse osteotomy model lacking mature T- and B-cells the mineralization process was accelerated[29]. However, in these animals an imbalance between collagen subunits I A1 and I A2 was observed. Histologically, the resulting arrangement of collagen I fibers appeared porous and osteoblasts showed an altered distribution within the fracture callus. These changes within the mineralization process were attributed to the lack of T-cells, using mouse models with either B- or T-cell deficiency[29]. Thus, T cells guide osteoblast distribution and the deposition of collagen I during callus formation. Later, homeostasis of bone is mediated mainly by the interaction between osteoblasts and osteoclasts, which derive from monocytes. The balance between these cell types is tightly regulated by the response of the respective progenitors, as well as T-cells acting during the remodeling phase of fracture healing[29].
The participation of each of the described cell types is required for successful fracture healing. In the following sections, the interplay between different immune cell types and osteoprogenitor cells will be more closely elaborated, starting with the cells of the innate immune response followed by the cells of the adaptive immune response, and the complement system as a mediator between them. Finally, a brief overview on the challenges of preclinical testing of immune-based therapeutic strategies to support fracture healing will be provided.
ROLE OF THE INNATE IMMUNE RESPONSE IN FRACTURE HEALING
Neutrophils
Neutrophils represent an essential part of the innate immune system. Neutrophils derive from common myeloid progenitor cells, which in turn originate from pluripotent hematopoietic stem cells (HSCs) in the bone marrow. The recruitment of neutrophils to the fracture hematoma may be mediated by released DAMPs originating from injured cells at fracture sites[16]. Mitochondrial particles can act as DAMPs[30], and cytokines such as IL-1α or IL-8 (also known as CXCL8) are known to recruit neutrophils to injury sites[31,32]. With regard to the fracture healing process, neutrophils have been described to strongly induce the initial inflammatory reaction (Figure 2), but their role in the actual healing process is rather underrated.
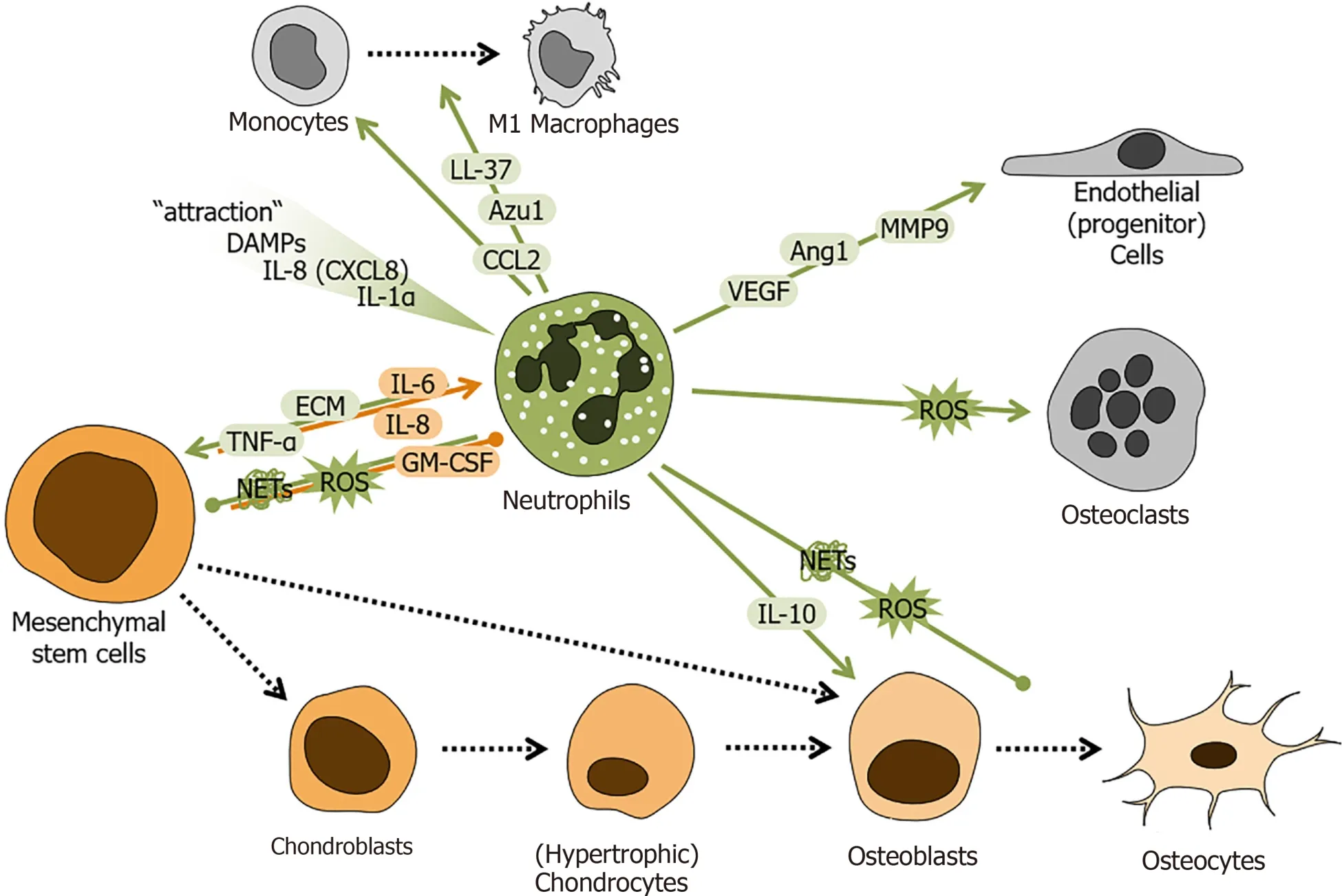
Figure 2 Schematic overview of the regulatory role of neutrophils during fracture healing.
In vivo, depletion of neutrophils is a common method of investigating their effect on healing processes. In small rodents like mice and rats, neutrophil depletion was shown to delay fracture healing[15], with increased stiffness of the newly formed bone[33], possibly due to reduced MSC infiltration and favored chondrogenic differentiation[34]. This is supported byin vitrostudies, showing that neutrophils form a kind of extracellular matrix that supports MSC influx into the fracture hematoma[35], but also inhibit matrix formation by MSCs[36]. Furthermore, a negative effect of freshly recruited neutrophils was observed, which induced apoptosis of undifferentiated and differentiated osteogenic cells[37]. Here, a reactive oxygen species (ROS)-dependent mechanism was proposed. This is in contrast to a recent study, showing that neutrophils alter cytokine release by MSCs but not their osteogenic differentiation[38], suggesting that the activation status of the neutrophils is critical in this process.
The observed delay in fracture healing in neutrophil depleted mice was no longer observed when an additional thorax trauma was present, emphasizing the role of neutrophils in systemic inflammatory responses following trauma[15]. Also, the application of neutrophils directly after fracture was reported to improve bone healing in these neutrophil depleted mice. As a possible regulatory mechanism, CCL (C-Cmotif chemokine ligand) 2 induced recruitment of monocytes was discussed[39]. CCL2, LL-37 (cathelicidin), AZU1 (azurocidin, also known as cationic antimicrobial protein CAP37 or heparin-binding protein), and other neutrophil secreted factors may pave the way for inflammatory monocytes[40]. Therefore, an indirect role of neutrophils in the healing process is possible, as monocytes are known to support healing by a CCL2-dependent mechanism, which gives a differentiation signal to MSCs[41].
Further investigations suggest that neutrophils may support angiogenesis and revascularization during the healing process, by releasing factorse.g., VEGF (vascular endothelial growth factor) and Ang1 (Angiopoietin 1), inducing inflammatory angiogenesis[42,43]. In addition, neutrophils were shown to release MMP9 (matrix metalloproteinase 9) in response to VEGF, inducing angiogenesis in hypoxic tissue[44].
TNF-α release from neutrophils was also suggested to be a possible mechanism by which neutrophils accelerate wound healing[45,46] and lead to early strength of surgical incisions[47]. However, the role of MSCs was not investigated in these studies. In MSCs, TNF-α was reported to enhance migration[48] but also to inhibit osteogenesis[49,50]. Thus, the direct effect of neutrophils on MSC development remains controversial.
The neutrophil to lymphocyte ratio and neutrophil counts were established as tools to predict trauma outcome and as markers for complication development. In most studies, high circulating neutrophil counts were associated with a higher risk of complications or postoperative mortality[51-55]. Furthermore, a higher neutrophil count in the blood of trauma patients was identified as a predictor for the development of delayed bone healing[56]. In addition, it was shown that neutrophil depletion reduced sepsis development after chest trauma in mice[57]. A variety of studies suggest that neutrophils play a major role in severe complications after trauma such as acute respiratory distress syndrome (ARDS), systemic inflammatory response syndrome (SIRS), MODS (multi-organ dysfunction syndrome), or ischemiareperfusion damage[57,58]. MSCs were suggested as a possible tool to reduce/ prevent tissue damage in these situations. MSCs reduce apoptosis of neutrophilsin vitro[59] and cause reduced attachment of neutrophils to endothelial cells[60]. The application of MSCs improved phagocytosis in a bacterial sepsis model in mice[61], reduced infiltration of neutrophils into the liver[62], and consequently improved survival in both studies. Furthermore, MSCs reduced inflammation, neutrophil infiltration, and the release of tissue-harming factors by these cells in an injured gut model or a vasculitis model[63,64]. ROS release was either increased[65,66] or reduced[67] depending on the study, but in general, there is consensus that MSCs seem to balance the neutrophil response.
Apart from strong induction of overall inflammation, neutrophils have another defense mechanism, called NETs[68,69]. Consisting of released chromatin covered with antimicrobial peptides and proteases, NETs are strongly bactericidal[68]. There is evidence that NETs are formed in response to trauma[70,71]. However, a general pathologic role of NET formation in healing processes has not yet proven. Nevertheless, overshooting NET formation can lead to pathological[72] or delayed healing[73]. Furthermore, NETs are known to be toxic to epithelial and endothelial cells[74,75]; thus, a toxic effect on MSCs is feasible. However, one study showed reduced NET formation and increased survival in a mouse model of lipopolysaccharide (LPS)-induced acute lung injury due to MSC application[76].In vitrostimulation of neutrophils with MSC-conditioned medium increased NET formation, although the overall response was delayed[77].
Another aspect that needs to be considered when discussing neutrophils and trauma is neutrophil phenotypes. Neutrophils are directly affected by trauma. They exert reduced reactivity to stimuli after trauma like bacterial infections[78] or fMLP (N-formylmethionyl-leucyl-phenylalanine)[79-81]. Reduced chemotaxis can be observed to different cytokines after lung trauma[70]. Surface markers [e.g., cluster of differentiation (CD) 62 and 11b, or L-Selectin] and receptors (e.g., CXCR1, CXCR2, or FcγRII, also known as CD32) are altered after trauma and thus also influence neutrophils reaction to other stimuli (reviewed in [78]). Recently, the categorization of neutrophils into “N1” and “N2” phenotypes, similar to monocyte/macrophage and Tcell types, was proposed. Neutrophils were divided into inflammatory (N1) and antiinflammatory (N2)[82]. Two recent reviews reported the role of neutrophil phenotypes in septic complications after trauma[83,84]. However, their distinct role in tissue repair and their effect on MSCs remain to be elucidated.
In summary, a clear role of neutrophils in the induction of inflammation and possible complications such as SIRS and ARDS has been identified. Their role in the actual healing process and their effect on MSCs remain largely unclear and different results from studies indicate both positive and negative effects on fracture healing. The analysis of neutrophil phenotypes within these results could help to clarify the role of neutrophils in fracture healing. An overshooting inflammatory response and NET formation by neutrophils are, however, consistently related to tissue damage and prolonged healing.
Monocytes and macrophages
Monocytes and macrophages also belong to the innate immune system. Like neutrophils, systemic or circulating monocytes and macrophages derive from the common myeloid progenitor cells in the bone marrow. In many organs, including bone, tissue-resident macrophages also exist, which predominantly derive from embryonic macrophages. In bone, these tissue-resident macrophages are named osteomacs. Osteomacs are closely associated with areas of bone formation, forming a canopy-like structure on top of the active osteoblasts[85]. Upon depletion of macrophages in a macrophage-fas-induced apoptosis (MAFIA) mouse model, not only the osteomacs but also the layer of active osteoblasts were lost[86], suggesting that macrophages play an active role in osteoblast mediated bone formation and hence fracture healing[87]. Monocytes infiltrate the injury site, usually within one day following fracture. Depending on the milieu they differentiate into immunogenic M1 macrophages or immunosuppressive M2 macrophages, DCs or later even into bone resorbing osteoclasts (Figure 3). In the early inflammatory phase of fracture healing monocytes are stimulated towards the pro-inflammatory M1 phenotype by inflammatory cytokines and to a lesser extent by bacterial products, such as LPS. These M1 macrophages phagocytose cellular debris and pathogens, produce large amounts of nitric oxide and ROS, and secrete cytokines,e.g., interferon (IFN)-γ, TNF-α, and IL-6. Thus, M1 macrophages contribute to the pro-inflammatory response following fracture. The cytokines secreted by M1 macrophages support homing of MSCs to the site of fracture[85,88]. Transforming growth factor beta (TGF-β) and associated activation of NADPH (nicotinamide adenine dinucleotide phosphate) oxidase 4 (NOX4) and focal adhesion kinase seem to play a crucial role in this process[89]. Later, during the granulation phase, the phenotype switches towards anti-inflammatory M2 upon stimulation with IL-4 and IL-13. M2 macrophages, which primarily secrete IL-10, actively support tissue repair during soft and hard callus formation by suppressing inflammation[22,90]. IL-10 production, also associated with tolerogenic DCs and regulatory B-cell function, is critically required for fracture healing and bone health. IL-10-/-mice were reported to develop osteopenia in both cancellous and cortical bone by suppressing new bone formation[91].
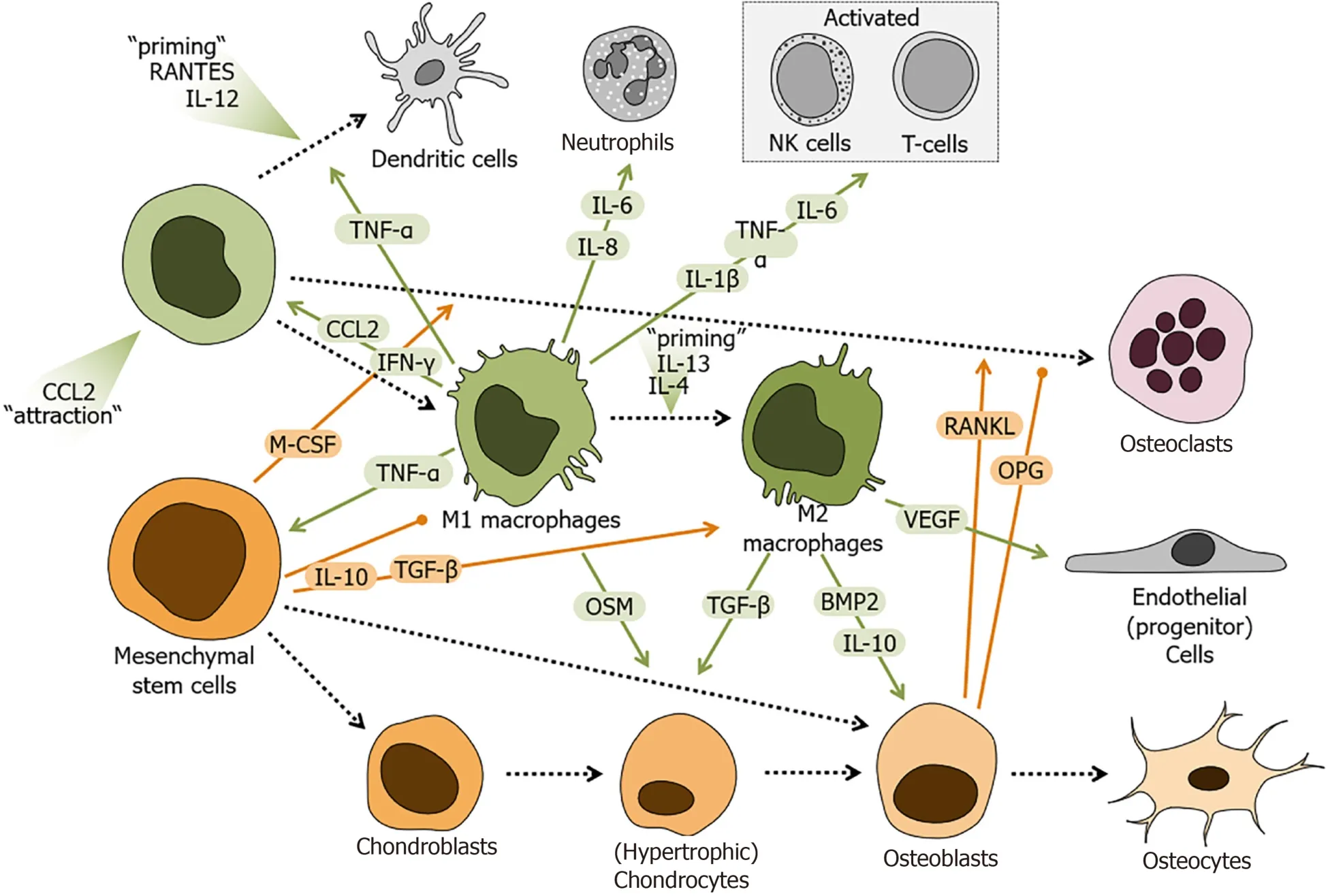
Figure 3 Schematic overview of the regulatory role of monocytes and macrophages during fracture healing.
In a mouse model of intramembranous bone formation, macrophages were closely associated with woven bone deposition by osteoblasts during all the phases of the healing process[92]. Depletion of macrophages affected deposition of woven bone and impaired healing of the defect in this model. Interestingly, macrophage depletion at the time of injury was more detrimental compared to depletion of macrophages at later stages of fracture healing[92]. This observation was confirmed in a mouse model investigating cancellous bone healing of drill holes[93]. In mouse models of endochondral fracture healing, depletion of macrophages in the early inflammatory phase of healing, resulted in reduced callus size, delayed hard callus formation, and delayed fracture union[22,94].
Somein vitrostudies have closely investigated the effect of the different macrophage phenotypes on bone formation. Conditioned medium from non-activated J774A.1 murine macrophages increases alkaline phosphatase (ALP) activity in MSCs, an effect mediated by secreted bone morphogenetic protein 2 (BMP2)[41]. This result was confirmed by another study showing that non-activated human monocytes enhance proliferation, ALP activity, and expression of osteocalcin and osteopontin, in human bone marrow derived MSCs (BMMSCs). It was found that these effects were not dependent on cell-cell-contacts but partially dependent on BMP2[95]. When cell-cellcontact is provided, non-activated human monocytes stimulate bone formation by BMMSCsviathe cytokine oncostatin M (OSM), which is secreted by monocytes directly upon cell-cell-contact with MSCs[96]. There are few studies that suggest that the observed effect is enhanced in M1 macrophages[97-99], and that this effect depends on OSM secreted by LPS challenged macrophages following activation of prostaglandin E2 (PGE2) and cyclooxygenase-2 (COX-2)[99,100].
When investigating the roles of monocytes and macrophages in fracture healing, the individual age has to be considered. Comparing fracture healing in young (3 mo) and old (24 mo) mice, no difference in the amount of macrophages in the fracture callus was observed. However, a constant up-regulation of M1/pro-inflammatory gene expression was observed in the macrophages of old mice. Therefore, preventing the infiltration of macrophages into the fracture site improved healing outcomes in old mice[101,102]. Similarly, when surgical reposition of the fracture is required, anesthetics may affect MSC-macrophage interaction. For example, local anesthetics,e.g., lidocaine and bupivacaine, have been reported to directly inhibit secretion of proinflammatory cytokines by macrophages, without affecting their viability[103]. Furthermore, these drugs may alter MSC effects in macrophage polarization by attenuating TNF-α and PGE2 secretion[103].
MSCs have been shown to suppress the M1 phenotype in macrophages, including the associated secretion of pro-inflammatory cytokines, in favor of the M2 phenotype with increased production of anti-inflammatory cytokines. This phenotypic switch from M1 to M2 macrophages is possibly mediated by PGE2[104-106],viaIL1RA (IL-1 receptor alpha), and IL-6[107], and/or by activation of NF-kB (nuclear factor-kappa B) and STAT-3 (signal transducer and activator of transcription 3), which is thought to require IFN-γ mediated indoleamine 2,3-dioxygenase activation[108,109]. Additionally, MSCs seemed to reduce CD86 and major histocompatibility complex (MHC) class II expression in LPS-stimulated macrophages, impairing their immunogenic effects on CD4+T-cell[104].
MCs
Although, best known for promoting allergic reactions[110], MCs also actively participate in fracture healing[20]. MCs derive from hematopoietic progenitors in the bone marrow and characteristically express CD34 and the surface marker c-Kit (protooncogene c-KIT, also known as CD117) that is important for MC growth, differentiation and survival[111]. MC progenitors are released from the bone marrow into the blood stream and finally mature in the mucosal or connective target tissues dependent on the local environment and growth factor availability[112]. In their large number of secretory granules, MCs store numerous preformed mediators, including cytokines and chemokines (e.g., IL-1β, IL-6, IL-8, TNF-a), histamine, heparin, enzymes (e.g., tryptases, chymases), and growth factors [e.g., VEGF, fibroblast growth factor (FGF), TGF-β]. These mediators can be rapidly released upon activation by cytokines, complement factors, or immunoglobulins (Ig),i.e., through crosslinking of the FceRI (Fc epsilon RI) receptorviaIgE[113]. MCs interact with many other immune cellsviathese mediators, thus, contributing to both the innate and adaptive immune responses[114]. Furthermore, MCs are capable ofde novosynthesis of several mediators (e.g., IL-1, IFN-g, RANKL). Many of these MC mediators are known to exert osteo-catabolic (RANKL, TNF-a, histamines) or osteo-anabolic effects [TGF-β, FGF, GM-CSF (granulocyte-macrophage colony-stimulating factor)][115], which is why MCs are supposed to regulate bone metabolism (Figure 4). Confirming this, patients with postmenopausal osteoporosis, rheumatoid arthritis, osteoarthritis, or other inflammatory diseases affecting bone, display increased numbers of MCs in the bone marrow[116-119].

Figure 4 Schematic overview of the regulatory role of mast cells during fracture healing.
Experimental phenomenological studies showed that MCs may also play a role in bone fracture healing. They are present in the fracture callus, especially in the early soft callus near blood vessels and in the later bony callus in proximity to osteoclasts[120,121]. Recent more mechanistic studies in various MC-deficient mouse models revealed the roles of MCs in fracture-induced inflammation, angiogenesis, as well as in anabolic and catabolic activities during the healing and remodeling process. In more detail, bone healing was delayed in MC-deficient KitW-sh/W-shmice indicated by an impaired transformation of woven bone into lamellar bone, reduced revascularization and increased osteoclast parameters[122]. In MC-deficient Cpa3Cre/+mice, bone repair was also impaired shown by reduced vascularization, bone mineralization and cortical bridging of the fracture callus[123]. However, these mouse models have the drawback that c-Kit is also expressed in osteoclasts and some immune cells, and Cpa3 (carboxypeptidase A3) in basophils and T-cells. Overcoming these limitations, fracture healing was investigated in MC-deficient Mcpt5 Cre R-DTA mice that lack connective tissue MCs without affecting other cell populations[124]. Interestingly, the levels of pro-inflammatory cytokines including IL-6 or CXCL1 were significantly reduced after fracture both systemically and locally in these mice. In addition, chemokine concentrations of KC (keratinocytes-derived chemokine, also known as CXCL1), MIP-2 (macrophage inflammatory protein 2, also known as CXCL2) and granulocyte colonystimulating factor (G-CSF), known to attract neutrophils, were significantly reduced in MC-deficient mice. This confirmed that fewer neutrophils and macrophages were recruited to the fracture hematoma. These results indicate an important role for MCs in fracture-induced local and systemic inflammation by the release of inflammatory mediators inducing the recruitment and activation of immune cells. Later in healing, callus bone content was increased in MC-deficient Mcpt5 Cre R-DTA mice associated with reduced osteoclast numbers, indicating that MCs may enhance osteoclast activity during callus remodeling. Supporting these findings,in vitroanalysis further showed that MCs promote osteoclastogenesisviagranular mediators, especiallyviahistamine[124].
In conclusion, MCs are present during the whole fracture healing process and mainly modulate the inflammatory response, vascularization and osteoclastic bone remodeling by using their broad spectrum of mediators. Hence, MCs can also influence MSCs present during the healing process as MSCs are responsive for MC mediators through various receptors,e.g., IL-1 receptor (IL-1R), IL-6R, TNFR (TNF receptor), CXCR1, TGFβRI (TGF-β receptor 1), or bFGFR (basic FGF receptor)[125]. On the other hand, MSCs also secrete factors including TGF-β, VEGF, or IL-6[126], that could modulate the function of MCs, as they express the appropriate receptors (TGFβ R1/2, VEGF receptor, IL-6R)[127]. MSCs are recruited to the injured region following fracture and initiate the repair phase by differentiation into chondrocytes and osteoblasts[128]. A direct interaction of MCs and MSCs during fracture healing has not been identified so far. In physiological bone turnover of MC-deficient KitW/W-vand KitW-sh/W-shmice, osteoblast parameters were changed probably due to an altered MSC differentiation capacity[129,130]; however, the underlying mechanisms need to be further investigated.In vitro, several studies observed effects of MCs on MSCs orvice versa. Co-culture experiments revealed that MCs can promote the proliferation of MSCs, but inhibit their differentiationviathe PDGF (platelet-derived growth factor) pathway[131]. Furthermore, pre-incubation of MSCs with exosomes isolated from MCs induced the migration of MCSsviaexosomal TGF-β[132]. Pre-treatment of MSCs with MC conditioned medium improved the therapeutic effect of MSCs in atopic dermatitis in mice[133].Vice versa, MSCs can also influence MCs in a co-culture system, by reducing their degranulation and cytokine production[134]. Moreover, the culture medium of MSCs pre-treated with TNF-a inhibited MC activation and histamine release in a model of allergic conjunctivitis[135]. MSC administration in various inflammatory settings including interstitial cystitis[136], atopic dermatitis[137], intracranial aneurysm[138], osteoarthritis[139], or allergic rhinitis[140], improved disease outcome,i.e., by reducing the number of MCs or their degranulation.
Hence, the effects of MCs on MSCs during bone healing are likely due to influencing migration, proliferation, and differentiation of MSCs. This might be of special interest in conditions of MC accumulation as observed in osteoporosis and probably also during fracture healing in osteoporotic bone or other inflammatory conditions in bone. Moreover, MSCs might also directly influence MC behavior during fracture healing by modulating MC numbers, degranulation, cytokine production and mediator release. In conclusion, more and more specific roles of MCs in fracture healing have been identified in recent years; however, the crosstalk of MCs and MSCs in this context requires further elucidation.
DCs
DCs derive from common myeloid progenitor cells in the bone marrow. DCs are specialized antigen presenting cells (APCs) that can also take up and process antigens, and have the capacity to stimulate resting T-cells in the primary immune response[141]. DCs process phagocytosed antigens into peptides in order to present them to TcellsviaMHC molecules on their cell surface, which primes T-cells as part of the adaptive immune response[142]. DCs differentiate from monocytes and secrete IL-12, favoring the differentiation of naïve CD4+T-cells toward T helper type 1 (Th1) cells, thus, contributing to the pro-inflammatory response required for homing of MSCs. Therefore, DCs are assumed to be active mainly in the early phases of fracture healing (Figure 5). However, the specific roles of DCs in fracture healing are yet to be elucidated. MSCs in turn have been shown to impair the maturation of DCs from monocytes or CD34+hematopoietic precursors[143]. As a result, fewer pro-inflammatory cytokines were secreted. This MSC mediated inhibition of DC function seems to be dependent on cell-to-cell contacts[144]. Another study suggested that MSCs inhibitory effects on DCs are related to the production of TGF-β and downregulation of DC costimulatory molecules (e.g., CD40, CD80, CD86), thus contributing to the activation of regulatory T-cells (Tregs)[145,146]. Yet another study suggested that MSCs secrete growth-regulated oncogenic chemokines when co-cultured with monocyte-derived DCs, which acquire a myeloid-derived suppressor cell-like phenotype under this condition[147]. Furthermore, MSCs were reported to induce expression ofSOCS1(suppressor of cytokine signaling 1) in DCs in an IL-6-dependent manner. This way, DCs acquire a tolerogenic phenotype with increased production of IL-10[148].
NK cells
NK cells, as part of the innate immune system, make up approximately 5% to 10% of all lymphocytes within peripheral blood. They derive from common lymphoid progenitor cells, which originate from HSCs in the bone marrow. Upon their primary mode of action, conventional NK cell subsets can be characterized by the expression of surface marker CD56. CD56dimNK cells mainly exert cytotoxic activities against tumor or infected cellsviaa MHC class I dependent recognition mechanism. CD56brightNK cells show an increased cytokine production capacity mainly secreting IFN-γ and TNFα, thereby, amplifying immune responses[149,150]. NK cells require priming by IL-12, IL-15, IL-18, IL-21, and IFN-α/β prior to activation which underlines that their range of action is widely believed to be within infectious environments, where classically activated NK cells contribute to the Th1 response[151-153].
The role of NK cells during trauma and their interaction with MSCs is not fully understood and seems strongly dependent on the current inflammatory status. After trauma, NK cells are among the first cells to arrive at the site of injury attracted by TNF-α and IL-6[21] (Figure 5). General immune suppression as a response to major trauma also affects NK cells. For example IFN-γ secretion was suppressed, following major trauma, when facing infectious challenges mimicked by Staphylococcus aureus[154].In vitro, NK cell activity was shown to be suppressed when incubated with fluids from early fractures or soft tissue injuries[155]. Decreased phosphorylation of mTOR (mechanistic target of rapamycin) and increased CD117 expression were identified as regulators of trauma-induced NK cell dysregulation[156]. To escape NK cell-driven lysis due to generally low MHC class I expression or NK cell mediated harm through massive IFN-γ production, MSCs were shown to be able to adapt within inflammatory environments. For example, MSCs were reported to increase MHC class I expression in response to high IFN-γ levels or increased resistance against cytotoxic NK cells upon Toll-like receptor 3 stimulation[157-159]. MSCs were found to be recruited to non-infectious environments, by CXCL7 secreted by primary unstimulated NK cells[160]. Classically, MSCs are reported to exhibit immune-suppressive properties towards NK cells — they secrete IL-10, TGF-β, and PGE-2, thereby limiting NK cell function and proliferation[161-163]. However, the immune-stimulatory effects of MSCs have also been reported. CD56brightNK cells, primed with IL-12 and IL-18, showed increased secretion of IFN-γ when co-cultured with MSCs without direct cellcell contact[164]. CCL2 was identified as the main immunomodulatory cytokine in this process. MSCs secreted CCL2 in response to IFN-γ, which primed NK cells for additional IFN-γ release in a positive feedback loop[165]. A recent study showed a time-dependent effect of MSCs on NK cells in the context of infected tissue injury. Shortly after injury (4 h) MSCs induced a pro-inflammatory response in NK cells by stimulating IFN-γ release. However, 24 h post-infection, MSCs induced a senescenceassociated NK cell phenotype (SASP) by TGF-β and IL-6 secretion, which was accompanied by a change in the population from CD56brightCD16+to CD56brightCD16-. SASP NK cells then triggered further IL-6 release, angiogenesis, and MSC proliferation, overall favoring tissue regeneration[166].
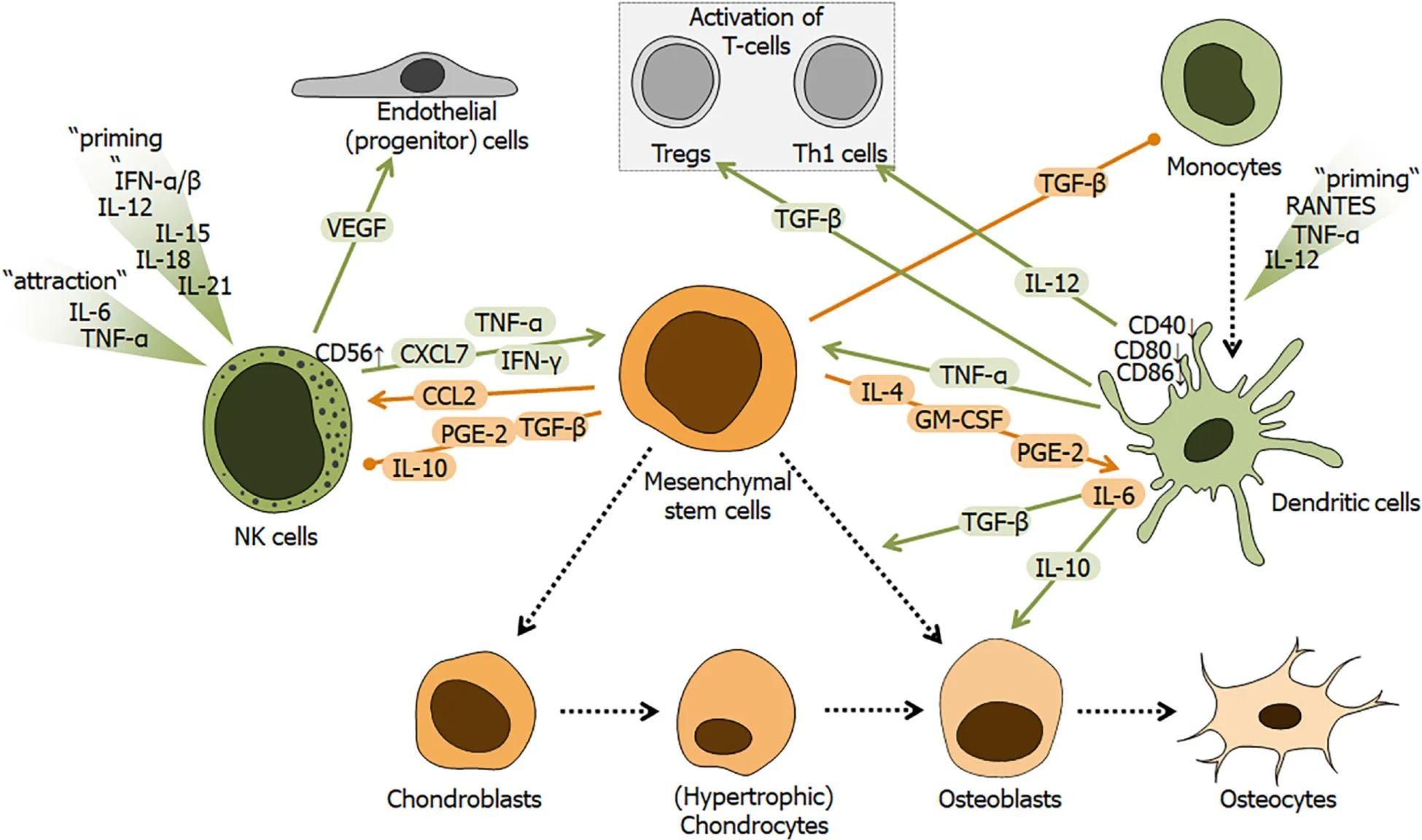
Figure 5 Schematic overview of the regulatory role of dendritic cells and natural killer cells during fracture healing.
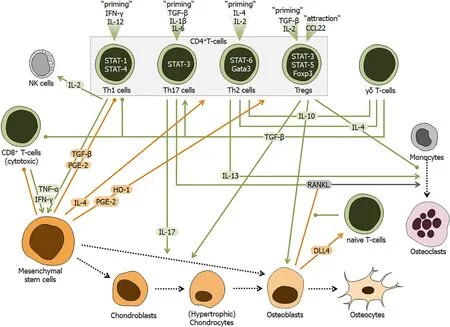
Figure 6 Schematic overview of the regulatory role of T-cells during fracture healing.
In summary, these studies suggest, that there is a tight interplay between NK cells and MSCs during fracture healing. However, the effects strongly depend on the inflammatory status.
ROLE OF THE ADAPTIVE IMMUNE RESPONSE IN FRACTURE HEALING
T-cells
T-cells play crucial roles in the adaptive immune response. They are of hematopoietic origin, as they derive from common lymphoid progenitor cells in the bone marrow. Maintenance of these common lymphoid progenitor cells is strongly dependent on the recently described subset of osteolectin positive MSCs, which can be found in close proximity to the arterioles in the central bone marrow and the endosteum[4]. It has been shown, that these osteolectin positive MSCs are required for T-cell as well as Bcell mediated bacterial clearance following an infection[4]. Using bone regeneration as a model, the effect of the adaptive immune system and more specifically the impact of T-cells has been investigated in order to first understand their role in regenerative processes and to secondly explore the possibility to use immune modulatory strategies to develop new therapeutic approaches. Similar to the cells of the innate immune response, T cells are also involved in many steps of fracture healing[167] (Figure 6). However, their activation status seems to be crucial. As discussed in the introduction, there is a loss of regenerative capacity in mammals starting after birth and upon aging. At birth, the adaptive immune system is still naïve. The number of naïve T-cells decreases with age, changing into effector, effector/central memory and terminally differentiated T-cells in a rate that is dependent on the antigens the individual encounters over time[168]. This highly individual immune aging process is therefore somewhat separated from chronological aging. In order to determine whether immune age influences bone properties, an approach was chosen where one group of mice was aged under sterile (specific pathogen free) conditions while a complementary group was housed under conditions where they encountered environmental pathogens. While the first group remained more or less immunologically naïve over the period of two years, the second group gained immune experience, developing central and effector memory cells[169]. Comparing bone parameters from these two groups of mice showed a stiffer and more brittle bone in animals with an experienced/aged immune system[169]. This is an indicator of the negative effect the change from the naïve immune status towards an experienced immune status, with central and effector memory T-cells and terminally differentiated T-cells, has on bone. Also the lack of Tcells, especially naïve T-cells, results in lower bone quality and delayed bone healing[169,170]. The above mentioned osteolectin positive MSCs, required for maintenance of the common lymphoid progenitor cells and adequate T-cell response, have been reported to decrease in number with age[4,5]. As the number of naïve T-cells also decreases with age, a direct correlation between these two cell types may be feasible. However, the effects of the MSCs on the T-cells seem to be strongly dependent on the cells activation status. It has been proposed that MSCs cause a downregulation of Fas receptor and Fas ligand on T-cell surface and thus, may rescue T-cells from activationinduced cell death[171]. TGF-β and hepatocyte growth factor, secreted by MSCs, have been identified to be soluble mediators suppressing T-cell proliferation, a process that can be augmented by cell-cell-contacts between the two cell types[172]. An immune composition with high levels of effector memory and terminally differentiated T-cells could thus be an indicator of delayed or disturbed bone healing. Indeed, a clinical study showed that delayed bone healing occurred in patients with high levels of terminally differentiated CD8+T-cells[170]. Thus, high percentages of terminally differentiated CD8+T-cells in peripheral blood could represent a biomarker for delayed healing that could easily be identified within one hour after a patient is hospitalized with a bone injury, opening possibilities for early intervention[170]. Amazingly, high percentages of these effector T-cells in peripheral blood or within the fracture hematoma, were not a result of the injury, but a result of antigen exposure over time.
Better characterization of the different T-cell subsets during bone healing, may shed light on their role in the healing process. Recently, IL-17 producing γδ T-cells, which are present in the fracture during callus formation, have been identified to promote bone healing[173]. However, T-cell differentiation is also critically dependent on osteoblasts, which produce notch ligand DLL4 (delta like ligand 4), the key regulator for this process[174]. Thus, sepsis-induced ablation of osteoblasts contributes to an immune deficiency[175].
Tregs, described to play a pivotal role in peripheral immune tolerance, are able to modulate both the innate and adaptive immune responses. Heme oxygenase-1 (HO-1) is a key contributor to MSC-mediated suppression of allo-activated T-cells, and induction of Tregs. For example, MSCs induce, in a HO-1-dependent fashion, IL-10+Tr1 (T regulatory type 1) and TGF-β+Th3 Treg-subsets in allo- and T-cell receptoractivated lymphocytes[176]. Furthermore, HO-1 facilitates MSCs to induce Tregs from naïve T-cells and promote their proliferation[176]. Tregs have the ability to alter and kill target cells such as APCs and effector T-cells. Furthermore, they may influence inflammatory cytokine environments and metabolic pathways[177]. Thus, these cells are required for maintenance of self-tolerance, or preventing excessive inflammation and autoimmune diseases. In the setting of trauma, Tregs become prominent when the granulation tissue is formed and remain at the fracture site until remodeling starts. They contribute to the specific release of anti-inflammatory cytokines such as IL-10, inducing a shift towards a Th2 lymphocyte-mediated response and/or lymphocyte anergy, and thus, to profound (post-traumatic) immunosuppression[178,179]. Later during fracture healing, Tregs are thought to control osteoblast and osteoclast function.
In vivo, the number of Tregs in peripheral blood is inversely correlated to serum markers of bone resorption, not only in rheumatoid arthritis patients but also in healthy controls, suggesting that Tregs control bone destruction[180]. Increasing numbers of Tregs improved clinical signs of rheumatoid arthritis and suppressed local and systemic bone destruction[180]. Furthermore, the suppressive effects of Tregs on osteoclast differentiation were confirmedin vitro[180]. It was suggested that enhancing the activity of Tregs may beneficially influence the treatment of inflammation-induced bone loss observed in rheumatoid arthritis. Yet, the effects and regulatory mechanisms of Tregs on osteoclastogenesis were investigated only in a limited number of studies. In a monocyte and Tregs co-culture system, Tregs inhibited osteoclast differentiation and reduced the resorbed areas[181]. The authors have shown that this suppression of osteoclast differentiation was cytokine-dependent, as osteoclast differentiation was blocked by anti-TGF-β or anti-IL-4 antibody treatment. As no direct cell-to-cell contacts were required to inhibit osteoclast function by Tregs, TGF-β and IL-4 may represent the key cytokines for this suppressive function of Tregs[181].
RANKL promoting the differentiation of bone-resorbing osteoclasts is thought to negatively impact bone healing, but is also active in T-cell differentiation and proliferation[1]. In a study, patients with isolated closed tibial fracture were subdivided into normal healing and delayed healing groups, based on their healing progression. In these patients, CD45RA-CD62L-effector memory cells most effectively suppressed RANKL. These cells were present at lower frequencies and with functional impairment in patients with delayed healing[182]. Hence, bone-resorbing osteoclast formation may be favored in these patients, suggesting a possible mechanism for delayed bone healing[182]. Another study supported the findings that multiple reductions in Tregs function in delayed healing patients could produce long-lasting consequences in the bone fracture healing process[183].
It has become evident that tissue destruction is associated with a decrease in local regulatory processes, including a decrease in forkhead box P3 (Foxp3)-expressing Tregs. CCL22 is a known chemoattractant for Tregs. With the help of a controlled release system, composed of a degradable polymer with a proven track record of clinical translation, poly(lactic-co-glycolic) acid, capable of generating a steady release of CCL22 from a point source effectively recruited Tregs to the site of injection[184]. Upon administration of CCL22 in murine experimental periodontitis, increases in Treg-associated anti-inflammatory molecules, a decrease in pro-inflammatory cytokines, and a marked reduction in alveolar bone resorption were observed[184]. In addition, the application of CCL22 reduced clinical measures of inflammation and improved alveolar bone loss in a ligature-induced periodontitis in beagle dogs[184]. Thus, Tregs recruited to the site of injury by CCL22 are associated with a decrease in bone resorption by reducing inflammation. STAT-3 as a key signaling protein in the skeletal and immune system, and may be a key regulator in this process[185]. The study provides evidence that STAT-3 enhances Tregs-mediated suppression of counteracting inflammation, suggesting that STAT-3 could be used as a prognostic marker to identify patients at risk of developing delayed union or nonunion[185].
In another study, systemic infusion of MSCs improved cell-based bone regenerationviaupregulation of Tregs[186]. In this study, the immunomodulatory function of BMMSCs was provenin vitro. Systemic infusion of these BMMSCs significantly improved cell-based repair of critical-sized calvarial defects in a murine model[186]. In the implantation sites, IFN-γ and TNF-α levels were reducedviaupregulation of Tregs, resulting in marked enhancement of cell-based bone regeneration, but with only limited contribution of BMMSC homing[186].
Apparently, Tregs also contribute to impaired bone healing induced by local accumulation of CD8+effector T-cells (TEFF). The endogenous regeneration is impaired by increasing the primary "useful" inflammation toward a damaging level with Tregs regulating the pro-inflammatory reaction to enhance healing[187]. The study provided evidence that CD4+Tregs might counteract undesired effects of CD8+TEFF, as the healing outcome was improved by an adoptive Tregs therapy[187]. The data from the mouse osteotomy model were supported by clinical data showing that patients with impaired fracture healing have demonstrated higher TEFF/Tregs ratios compared to uneventful healers[187]. These findings demonstrated the key-role of a balanced TEFF/Tregs response following injury required for successful bone regeneration[187].
Although more and more studies show possible roles of the adaptive immune system in bone healing, the underlying mechanisms and involved cell types are still unclear and remain to be elucidated in further studies.
B-cells
Like T-cells, antibody producing B-cells belong to the adaptive immune response. They also differentiate from common lymphoid progenitor cells, which are derived from HSCs in the bone marrow. MSCs support the development of T- and B-cells from HSCs by soluble factors and cell-cell-contacts[188]. Both cell types infiltrate the fracture callus in a two-waved fashion (Figure 7). Interestingly, the number of B-cells seemed to exceed the T-cells during the fracture healing process, where B-cells progressively underwent effector maturation[189]. Early during callus formation Bcells have been described to undergo direct cell-cell-contact with osteoprogenitor cells, presumably regulating their differentiation. However, as described above, the lack of mature T- and B-cells accelerated the formation of the mineralized matrix in a mouse osteotomy model[29]. However, the observed changes within the mineralized matrix where attributed to the lack of T-cells, using mouse models with either B- or T-cell deficiency[29]. Another study suggested, that B-cells regulatory function is required for successful bone healing, as in patients with delayed healing of tibia fractures B-cells seemed to lose their capability to produce IL-10 with time[190]. Another study even suggested, that the initial expression of IL-10 by B-cells is diminished in patients developing a non-union[191]. During the healing process, these antibody and IL-10 producing CD27+B-cells effectively suppressed IFN-γ, TNF-α, and IL-2 expression in CD4+T-cells, as well as IFN-γ and TNF-α expression in CD8+T-cells in a Foxp3 dependent manner[191]. Likewise,in vitroCD19+CD27brightB-cells suppressed proliferation of CD4+T-cells and enhanced Foxp3 expression in Tregs. However, the mechanism was not dependent on IL-10 but TGF-β and direct cell-cell-contact[192]. Furthermore, CD19+CD27brightB-cells were reported to reduce the numbers of proinflammatory Th17 cells, independently of cell-cell-contacts[193]. This indicates that Bcells have crucial immunomodulatory roles during fracture healing.

Figure 7 Schematic overview of the regulatory role of B-cells during fracture healing.
However, B-cells may also affect bone cells andvice versa. For example, in rheumatoid arthritis patients, B-cells have been reported to suppress osteogenesisviaTNFα and CCL3[194]. Similarly, B-cells inhibited osteoblast maturation when challenged with G-CSF during homing of hematopoietic stem and progenitor cells. Simultaneous activation of osteoclasts, however, was attributed to T-cells[195]. Interestingly, Rag1-/-mice displayed higher than normal levels of osteoclasts, although lacking T- and Bcells[196]. As a possible explanation for this, a response to the elevated osteoblast function was suggested. In turn, MSCs may inhibit proliferation, activation, and antibody secretion of B-cells, possibly by altering MAPK (mitogen-activated protein kinase) activity[197]. It is assumed that MSCs inhibit B-cell proliferation by secreted factors inducing cell cycle arrest in the G0/G1 phase[198], and preventing B-cells maturation by inducing expression of maturation protein-1[199]. Furthermore, MSCs may suppress both B- and T-cell activation by secreted IFN-γ and cell-to-cell contactviaprogrammed cell death 1 receptor (PD-1) and its ligand (PD-L1)[200].In vitro, however, contradictory data exist, showing that MSCs may promote the proliferation and differentiation of B-cells[201].
COMPLEMENT SYSTEM
The complement system represents the major fluid phase innate immune surveillance system[202]. As a protein cascade, the complement system contains multiple serine proteases, which can be activated by different pathways early after trauma and during systemic inflammation[202,203]. The generated complement activation products can function as anaphylatoxins (C3a, C5a), opsonins (C3b), or membrane attack complexes (MAC formed by C5b-9) all of which help sensing and clearing of tissue debris, damaged cells, and pathogens after trauma[204]. However, if excessively produced or out of control by suppressed complement regulatory proteins (CRegs), activated complement may also reveal harmful features for the host[203].
On a cellular level, multipotent MSCs are critically involved in healing processes after tissue damage and bone fracture. A hypothesis-free global transcriptional analysis of the bone fracture region post trauma suggested, that several complement and coagulation factors are significantly upregulated at the fracture site[205]. Focusing on MSCs, it is well established that these cells express several key complement receptors (e.g., C3aR or C5a receptors) and membrane-bound CRegs (e.g., CD35, 46, 55, and 59) all of which play an important role in the concerted recruitment of leukocytes and subsequent induction of repair processes[206]. Trauma can also result in systemic complement activation with generation of the central anaphylatoxins C3a and C5a, which can induce all classical signs of inflammation. C3a represents a potent chemoattractant for MSCs[207] and C5a strongly chemo-attracts neutrophils and macrophages. Moreover, opsonisation of MSCsviaC3b deposition results in subsequent phagocytosis by macrophages. Concerning the terminal pathway, MAC formation on MSCs can lead to cellular lysis and thus impaired regeneration[206,208]. Concerning cell survival, generation of C5a can induce apoptosis of MSCs[209]; whereas, in contrast, it prevents apoptosis in neutrophilsviaenhanced expression of the anti-apoptotic Bcl-xL[210].
The multifaceted modulation of MSCs by complement activation products led to the development of novel therapeutic approaches. Nanoparticles spiked with the C5aR, functioning as a decoy mechanism for excessive C5a, revealed some protective effects for MSCs[211]. Similarly, “painting” MSCs with factor H, a central native complement inhibitor, turned the MSCs resistant against both a complement- and neutrophildriven attack[212]. Another approach addresses differentiation processes of MSCs: During the differentiation from MSCs to osteoblasts, C5aR upregulation is dependent on regulation of the urokinase receptor (uPAR) and downstream NfκB transcriptional program. Blocking the C5aR impaired osteogenic differentiation, indicating effective immune modulation of the MSC-driven regeneration process by targeted complement inhibition[213]. However, future translational studies need to investigate and prove the efficacy of such a complement-based MSC modulation.
IMMUNE CELL REGULATION A POSSIBILITY FOR PERSONALIZED TREATMENT TO SUPPORT FRACTURE HEALING?
Delayed or impaired fracture healing, which occurs in up to 20% of all fractures[214], and septic complications represent growing challenges in orthopedic and trauma surgery. Currently, failures in bone healing are detected radiologically 4-6 wk after the initial treatment. This considerably prolongs the healing time in patients with healing deficits. The above-mentioned studies describe crucial roles of the innate and adaptive immune system in these processes (Figure 8). Hence, immune scenarios characteristic in patients frequently developing delayed or impaired bone healing, or even septic complications were identified. The alterations in the immune response usually become apparent early in the healing process of a fracture, some even at the time of hospitalization of the patients. This opens new avenues for early interventions. With a tool to stratify patients with higher risks for delayed healing, therapeutic approaches to treat these patients are needed. With a demographically aging population the percentage of elderly patients, with an educated and aged immune system, presenting to the clinic with a fracture will rise. This factor has to be considered when developing and testing new therapeutic strategies based on immune modulation, emphasizing the need for a paradigm shift in animal models. In 2016, Badylak[215] stated that the immune system as a regulator of organ and tissue development and as an orchestrator of the healing process after injury is often neglected in research models. Although, there are rising numbers of studies emphasizing that the immune status of the pre-clinical model could be decisive for the research results[187,216,217], state of the art is to keep animals as clean as possible,i.e.most rodent model housing is often either pathogen free or specific pathogen free. In both housing conditions the immune experience is minimal. This is partly attributed to the fact that so far no standardized method has been found to measure or report immune competence or experience in pre-clinical models. Using mice with effector/memory T-cells, kept under housing conditions that allowed environmental pathogens[169,187], an immunomodulatory strategy was tested that enhanced cyclic adenosine monophosphate during the initial phase of bone healing through local application of the prostacyclin analogue Iloprost. Prostacyclin, previously used to treat bone-marrow edema[218,219], reduced the pro-inflammatory reaction of effector memory T-cellsin vitro, while strengthening the anti-inflammatory reaction of Tregs, one natural counterpart to CD8+effector cells[220]. In an immune experienced mouse osteotomy model local delayed release of Iloprost significantly enhanced bone healing, while reducing the number of CD8+T-cells in the early healing phases[220]. In contrast, a previous study reported that Iloprost inhibited bone healing in a rat fracture model[221]. The discrepancy in statements can be explained by the unstabilized fracture in the rat study or the systemicvslocal application of Iloprost. However, the immediate prostacyclin administration in the rat study probably prevented the necessary initial pro-inflammatory reaction and this caused the observed lack in healing. This demonstrates that the complexity of the bone healing process combined with the complexity of the immune system and reaction upon injury demands a very careful strategy when immunomodulation is to be achieved to improve bone healing. Especially as both systems, the bone and the immune system share signaling pathways. This means that by targeting one system one could very well also influence the other. Nevertheless, immunomodulation is a promising future treatment approach to enhance bone healing in patients with an overarching immune reaction to injury that will probably become a personalized therapy option, where the immune composition of the patient has to be taken into account. With bone being a model for regenerative healing, knowledge gained in bone research could become a blueprint to enable scar-less healing in non-regenerative organs in the future.
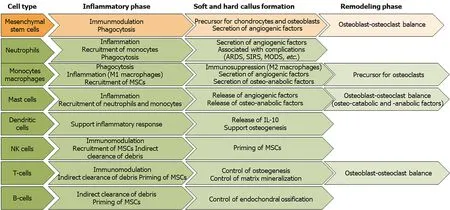
Figure 8 Overview of the roles of immune cells and mesenchymal stem cells during the different phases of fracture healing.
CONCLUSION
Bone is considered an osteoimmune system based on cooperatively acting bone and immune cells, cohabitating within the bone marrow. The different cell types are highly interdependent, sharing progenitors, mediators, and signaling pathways. MSCs with their manifold immunomodulatory and regenerative properties serve as progenitors for fibroblasts, chondrocytes and osteoblasts during fracture healing. Immune cells of the innate and adaptive immune system influence viability and differentiation capacity of MSCs during this process. Alterations in the immune response usually become apparent early in the fracture healing process, which opens new avenues for early interventions. However, to investigate new therapeutic strategies aiming to balance altered immune responses during fracture healing requires the address of not only the innate but also adaptive immune responses. This raises the need for advanced model systems.
杂志排行

World Journal of Stem Cells的其它文章
- Priming strategies for controlling stem cell fate: Applications and challenges in dental tissue regeneration
- Epigenetic regulation of dental pulp stem cells and its potential in regenerative endodontics
- Regulation of the mesenchymal stem cell fate by interleukin-17:Implications in osteogenic differentiation
- Why stem/progenitor cells lose their regenerative potential
- Nanofat: A therapeutic paradigm in regenerative medicine
- Application of adipose-derived stem cells in treating fibrosis