靶向EGFR蛋白降解剂及其在非小细胞肺癌中的应用
2021-12-23宋肖玲屈小娟曲思琪姜标
宋肖玲, 屈小娟,曲思琪,姜标
①上海科技大学 免疫化学研究所,上海 201210;②上海科技大学 生命科学与技术学院,上海 201210;③中国科学院上海有机化学研究所,上海 200032
1 EGFR基因突变与肿瘤靶向治疗
表皮生长因子受体(epidermal growth factor receptor, EGFR)是ERBB受体家族成员之一,属于酪氨酸激酶型受体(tyrosine kinase receptor),它是由28个外显子编码的1 186个氨基酸组成的170 kDa 的跨膜糖蛋白[1]。EGFR在细胞增殖与生存中发挥关键的控制作用[2]。EGFR激酶区突变是导致非小细胞肺癌(non-small cell lung cancer,NSCLC)癌变的主要原因之一[3-4],且具有EGFR突变的患者占非小细胞肺癌患者群体总数的10%~50%左右[4-8],因此EGFR是肺癌靶向治疗的一个关键靶点。该突变在亚洲、女性、非吸烟的非小细胞肺癌患者中较为多见[9]。EGFR突变多发生于EGFR激酶区,外显子19 LREA氨基酸缺失突变以及外显子21 L858位氨基酸的点突变(L858R)占据EGFR激酶区突变的87%,被称为常见突变[10]。此外,EGFR突变还包括外显子20重复/插入突变等其他类型突变。突变的EGFR与HER2的亲和力增强,更倾向于与HER2形成异源二聚体[11]。HER2与突变型EGFR结合后不需要配体EGF的刺激即可展示一定的磷酸化水平,同时下游信号通路也被一定程度地激活。伴随着EGFR突变体对信号通路的选择性激活以及EGFR蛋白较少的衰减引起信号通路的持续激活,细胞发生癌变[12]。
由于EGFR突变肺癌高度依赖EGFR蛋白激酶活性,特异性地抑制EGFR活性会杀死肿瘤细胞。第一代EGFR酪氨酸激酶抑制剂(tyrosine kinase inhibitor, TKI)的引入,对于部分携带EGFR敏感突变的NSCLC患者的治疗带来很大程度的改善。相比于传统化疗法,EGFR-TKI治疗在可接受的毒性范围内,明显延长无进展生存期[5,13]。但病人在接受EGFR-TKI治疗一段时间(8~16个月)后,都不可避免地出现耐药情况。研究发现导致NSCLC患者对第一代EGFR-TKI继发性耐药突变的因素错综复杂,但其中最主要的是EGFR第二突变T790M,占据所有因TKI治疗引起的获得性耐药NSCLC患者的60%左右[14]。2015年11月, 奥希替尼获得了FDA加速批准,用于经EGFR-TKI治疗时或治疗后疾病进展时出现的T790M突变阳性的NSCLC靶向药。奥希替尼的使用也会引起新的EGFR耐药突变的发生,其中EGFR C797S 第三突变占奥希替尼耐药患者的40%左右,但目前尚无有效化合物应对该困境。
由于传统小分子抑制剂类药物不可避免地会引发耐药现象,利用新的技术策略开发新型药物来治疗肺癌患者势在必行。EGFR突变的肺癌高度依赖于EGFR蛋白的激酶活性,而且发生耐药后近半数原因与EGFR蛋白的二次突变相关,因此利用蛋白靶向降解技术特异性地清除EGFR蛋白在肺癌治疗中具有很大的应用潜力。本文主要就EGFR靶向蛋白降解技术近年来的研究进展进行综述。
2 EGFR靶向蛋白降解剂的研究进展
目前,已有报道使用小分子抑制剂通过EGFR自身的降解机制来促进突变EGFR蛋白的降解,但治疗效果有限。例如,使用抑制剂PITSTOP2靶向抑制网格蛋白(clathrin)介导的内吞(CME)或用基因沉默技术敲低网格蛋白之后,会重新激活网格蛋白非依赖的内吞(CIE)途径,引导突变的EGFR至溶酶体被降解,同时EGFR信号通路被抑制,肿瘤细胞发生凋亡[15]。近期还有研究报道使用新型EGFR配体23-羟基白桦酸(23-hydroxybetulinic acid)衍生物——DPBA[16],通过促进 flotillin-1依赖的EGFR内吞,诱导EGFR在溶酶体中被降解,从而杀死肿瘤细胞。然而,由于这些方式多不具有很强的特异性,小分子发挥作用通常需要较高的浓度(如DPBA需要6µmol/L),其潜在的毒副作用比较明显。
由于干扰EGFR内源降解机制对突变EGFR的降解不具有特异性,通过人为导入其他降解机制来特异性地降解EGFR突变蛋白,被用来设计药物以求克服靶向药物的耐药问题。目前,多种蛋白降解系统已被人为改造,用来靶向性降解目的蛋白。按照作用机理的不同,靶向蛋白降解剂可以分为3类:①依赖泛素/蛋白酶体系统的降解剂——蛋白降解靶向嵌合体(proteolysis targeting chimeras, PROTACs);②依赖溶酶体发挥作用的降解剂——溶酶体靶向嵌合体(lysosome-targeting chimaera, LYTAC);③依赖自噬(macroautophagy/autophagy)途径发挥功能的降解剂。目前仅有前两种被报道用于EGFR的靶向降解。此外,本文还就利用订书肽来促进EGFR降解的技术一并进行综述。
2.1 靶向蛋白降解技术
PROTAC技术最早由Crews等人[17]于2001年提出。泛素/蛋白酶体系统是真核细胞调控细胞内蛋白水平的一种主要机制[18],该系统可以利用E3泛素连接酶给要被降解的蛋白打上特殊的泛素化标签,使其被蛋白酶体识别并最终被降解(图1)。利用该系统设计的PROTAC分子主要由3部分组成:①靶向目的蛋白的配体,目前多为靶蛋白的特异抑制剂;②E3泛素连接酶的配体;③与上述二者配体相连的连接子(linker)。这种双功能分子一端可以与靶蛋白结合,另一端可与E3泛素连接酶结合,通过将E3泛素连接酶特异地招募至靶蛋白附近对靶蛋白进行泛素化,并最终导致靶蛋白被26S蛋白酶体所降解[19-20](图1)。目前,PROTAC技术已成功地应用于多种靶蛋白的降解,其中针对核受体蛋白如AR、ER(estrogen receptor)等的蛋白降解剂已经开始进行临床试验,并初步取得了较好的抗癌效果。
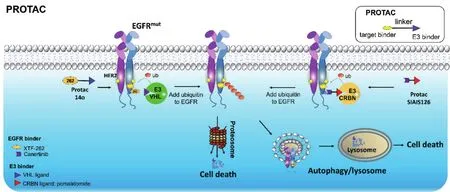
图1 靶向蛋白降解(PROTAC)技术。靶向蛋白降解剂是由3部分组成的双功能小分子:靶蛋白配体、E3泛素连接酶配体和连接子(linker)。突变的EGFR在体内倾向于与HER2形成异源二聚体并引起下游信号通路持续激活。加入蛋白降解剂14O(由XTF-262、VHL配体和连接子组成)和SIAIS126(由卡纳替尼、CRBN配体泊马度胺(pomalidomide)和连接子组成)后,蛋白降解剂会招募E3泛素连接酶VHL或者CRBN至EGFR,引起EGFR的泛素化,随后EGFR蛋白被蛋白酶体途径所降解。研究显示自噬溶酶体途径也参与降解剂SIAIS126对EGFR突变蛋白的降解
目前已有数篇文章报道了可以降解肺癌特异突变的EGFR 蛋白降解剂[21-25]。VHL和CRBN是蛋白降解剂中最常用的两种E3泛素连接酶。研究显示,用VHL和CRBN的配体来设计的蛋白降解剂均可很好地降解EGFR突变蛋白并发挥抗肿瘤作用。用来靶向EGFR的配体不仅有FDA已批准的EGFR靶向药如吉非替尼(gefitinib)、奥西替尼(osimertinib),还包含临床前在研的EGFR靶向抑制剂,它们均可用作EGFR的结合配体。EGFR蛋白降解剂因其EGFR靶向结合分子和连接子的不同对EGFR的降解能力各异。下面按照降解剂的作用效果来分别阐述EGFR降解剂的功能。
(1)可高效降解EGFR激活突变的EGFR蛋白降解剂
Ex19Del和L858R点突变分别占据了肺癌患者中具有EGFR突变人数的45%左右。目前报道的可以降解EGFRL858R的蛋白降解剂仅有3个:由美国西奈山伊坎医学院金坚教授团队开发的EGFR降解剂MS39和MS154[21]以及耶鲁大学Crews团队开发的Protac3[26]。这三个化合物均以吉非替尼为EGFR的靶向识别分子,在 HCC3255细胞中对EGFRL858R的半数降解剂量(DC50)介于3~25 nmol/L之间,这说明降解剂可以很好地杀死肿瘤细胞。
在已报道的可以降解EGFR Ex19Del的降解剂中,降解效果最强的是西安交通大学张三奇团队基于化合物F(compound F)和VHL配体开发的蛋白降解剂P3[27],它对HCC827细胞中EGFR ExDel的DC50低于1 nmol/L。其次是基于吉非替尼开发的EGFR降解剂MS39(VHL)、MS154 (CRBN)[21]和Protac3(VHL),它们在HCC827细胞中对EGFR Ex19Del的DC50分别为5、11和 11.7 nmol/L。EGFR蛋白降解剂在小鼠中有良好的耐受性,并且具有较好的药代动力学特征。MS39以50 mg/kg的剂量经静脉注射后在循环系统中可以实现较高的血药浓度——约5 µmol/L可维持8 h,且给药后24 h依旧可维持1 µmol/L左右的血药浓度。随后是张三奇团队基于自己研发的第四代EGFR TKI——Compound1,分别采用Lenalidomide和VHL-Ligand作为E3泛素连接酶的配体设计的蛋白降解剂degrader 2、degrader10[24]。这两个降解剂在HCC827对EGFRex19del的DC50分别为45.2、34.8 nmol/L。基于AZD9291和lenalidomide(CRBNLigand)开发的PROTAC 16c[22]对EGFRex19del的降解能力最弱,其在PC9细胞中的DC50仅为161 nmol/L,对该细胞的半数增殖抑制浓度(IC50)为413 nmol/L。
(2)可以降解EGFRL858R+T790M突变的蛋白降解剂
目前已报道的可以降解EGFRL858R+T790M突变蛋白的降解剂有基于VHL配体的降解剂14O和基于CRBN配体的降解剂SIAIS125/126。其中14O[25]是基于中国科学院大学谢婷团队研发的第三代EGFR 酪氨酸激酶抑制剂XTF-262开发的PROTAC,其对EGFRL858R+T790M的DC50为5.9 nmol/L,可以高效选择性地杀死H1975(EGFRL858R+T790M)细胞(IC50为19.3 nmol/L)。目前该降解剂尚未进行体内活性验证。
SIAIS125和SIAIS126[23]是上海科技大学姜标教授团队基于第二代EGFR TKI Canertinib和E3配体pomalidomide构建的由全碳链连接而成的2个PROTAC分子。这两个降解剂具有选择性降解EGFRex19del和EGFRL858R+T790M的能力,对EGFRL858R+T790M的DC50在30~50 nmol/L之间。这两个化合物在体外对肿瘤细胞具有良好的杀伤活性, 不仅可以阻滞细胞周期从而显著抑制H1975细胞增殖,还可以促进细胞凋亡,抑制细胞迁移和侵袭等。对其作用机制进行初步研究后发现,抑制泛素化途径可以显著抑制降解剂对EGFR蛋白的降解作用。同时,使用蛋白酶体和溶酶体抑制剂均不能完全阻断降解剂对EGFR蛋白的降解。降解剂处理细胞后,细胞的自噬加强。该研究首次展示了EGFR蛋白降解剂不仅可以通过蛋白酶体系统降解EGFR靶蛋白,同时还可以通过泛素/自噬溶酶体途径对靶蛋白进行降解[23]。
2.2 溶酶体靶向的EGFR降解
LYTAC技术最早由Bertozz教授团队[28]于2020年在Nature上报道。该技术利用细胞表面的溶酶体靶向受体(lysosome-targeting recepters,LTRs)可以特异性地识别带有某种特殊标记的靶蛋白,并定向地将其运送到溶酶体去降解的特性来发挥作用[29]。已报道的LTRs包括M6PR(mannose-6-phosphate receptor)[30]、LIMP2(lysosomal integral membrane protein type 2)和Sortilin等[31],其中CI-M6PR为介导溶酶体蛋白运输的主要受体[32-34]。将CI-M6PR的配体——寡糖肽基团(poly(M6Pn) polypeptides, M6Pn)修饰识别靶蛋白的抗体即可形成 LYTAC分子。
报道展示了可以降解EGFR靶蛋白的LYTAC分子叫Ab-2,由被寡糖肽特异修饰的西妥昔单抗(cetuximab-M6Pn glycopolypeptides)组成[28](图2)。Ab-2可以与CI-M6PR及EGFR形成复合物,将EGFR转运至溶酶体降解,而CI-M6PR返回细胞膜。研究发现,Ab-2只在CI-M6PR正常表达的细胞系中能够介导EGFR的溶酶体降解,而利用CRISPR技术敲除CI-M6PR后,降解效果消失。高浓度的M6P(5 mmol/L)可竞争性抑制Ab-2对EGFR的降解效果,同时采用溶酶体抑制剂氯喹(200 μmol/L)也可显著降低Ab-2的作用。作用时效性方面,10 nmol/L Ab-2处理3 h时即可诱导EGFR水平的降低,在12 h至24 h时达到最大降解程度(80%),对EGFR的降解效果可持续72 h以上。Ab-2也可引起乳腺癌细胞(BT474、MDAMB-361)和肝癌细胞(Hep3B、HepG2)中的EGFR降解。质谱法进行蛋白质组学分析,结果显示Ab-2处理显著降低EGFR水平,而CI-M6PR保持不变。这更进一步证明,在Ab-2诱导的EGFR降解过程中,CI-M6PR只起媒介作用。然而研究者发现,Ab-2诱导下还有其他蛋白水平发生变化,例如二氢蝶啶还原酶(dihydropteridine reductase)、核酸氧化损伤修复酶MTH1(NUDT1)及转录因子DP-1等蛋白的上调,引起变化的原因尚未被进一步阐明。

图2 溶酶体靶向降解技术。溶酶体靶向降解剂包含一个可以特异识别目的蛋白的抗体和CI-M6PR的配体寡糖肽基团。在细胞中加入溶酶体靶向降解剂后,该分子可以同时结合目的蛋白EGFR和CI-M6PR受体蛋白,并在CI-M6PR蛋白的介导下将目的蛋白EGFR转运至溶酶体发生降解
除了EGFR之外,LYTAC分子还可以成功降解细胞外或细胞膜上诱发疾病的分泌蛋白和膜蛋白,包括 CD71、PD-L1等[28]。LYTAC提供了一种新的针对胞外蛋白或膜蛋白的降解工具,进一步拓宽了靶向蛋白降解剂的适用谱。
2.3 订书肽介导的EGFR降解
订书肽(stapled-peptide)是一种经过化学修饰使得α螺旋中某两个氨基酸残基侧链形成共价连接,从而形成的具有稳定α螺旋结构的多肽链[35-37]。订书肽因其稳定的α螺旋结构、很好的细胞渗透性以及较高的抗水解能力,可以用来靶向胞内的蛋白-蛋白相互作用。利用订书肽来直接或间接招募E3泛素连接酶至EGFR并调控其降解,也是一种潜在有用的靶向EGFR降解的新策略。
2020年胡卓伟团队[38]首先报道了一种基于TRIB3-EGFR相互作用的订书肽SAH-JGZ4来靶向性降解EGFR(图3)。TRIB3与EGFR的相互作用可以调节肿瘤细胞EGFR的稳定性和肿瘤细胞干性。研究发现,TRIB3通过招募PKCα使得EGFR T654发生磷酸化,进而在E3连接酶WWP1作用下引起EGFR K689位发生泛素化(K63偶联方式)。这一泛素化可作为EGFR再循环的精确信号,使得EGFR再循环至细胞膜。基于上述原理,将EGFR胞内近膜区与TRIB3发生相互作用的关键区域(JM区)上第二个α螺旋进行化学改造后形成订书肽SAH-JGZ4。该订书肽与TRIB3有较高的亲和力,并具有很好的细胞膜透过性和胞内稳定性,可通过干扰TRIB3-EGFR相互作用加速EGFR降解,在细胞水平、CDX及PDX模型中都表现出显著的抗肿瘤活性,而且能够降低NSCLC细胞干性。靶向Protein-EGFR之间相互作用的新型药物分子的开发或许是EGFR驱动的癌症患者的新曙光。
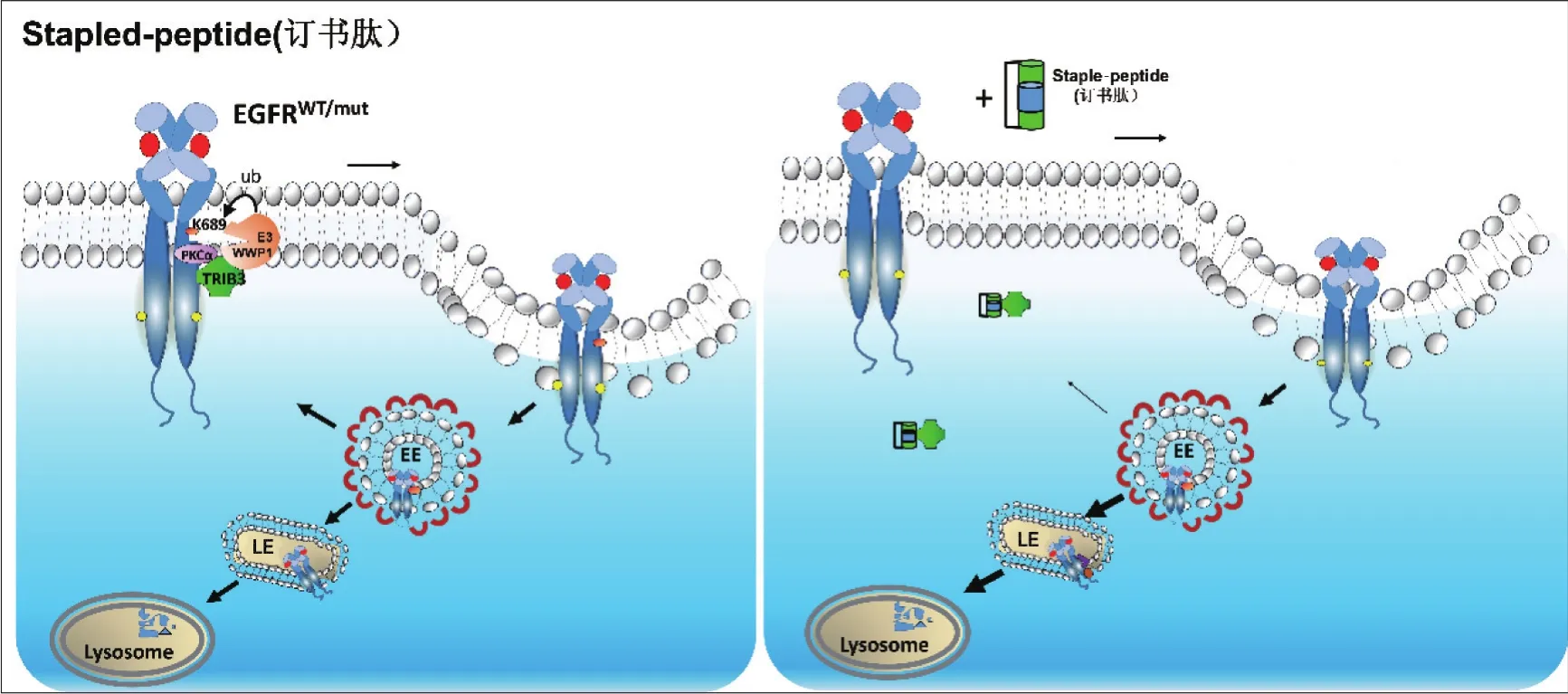
图3 订书肽介导EGFR降解的工作原理。订书肽SAH-JGZ4的氨基酸序列来自EGFR跨膜区与TRIB3蛋白特异相互作用的经过侧链修饰的α螺旋区,经过氨基酸侧链共价连接后所形成。左图:正常情况下,EGFR被激活后,会与TRIB3相互作用,并相继招募PKCα和E3泛素连接酶WWP1至EGFR,引起EGFR的K689位被泛素化。EGFR被内吞后,在早期内体被分选:具有该位点修饰的EGFR被循环至细胞膜表面继续发挥信号传导功能,不具有该位点泛素化修饰的EGFR被分选至溶酶体并降解。右图:在细胞中加入订书肽后,订书肽会干扰TRIB3与EGFR的结合,从而阻断了EGFR的K689位被泛素化。EGFR被内吞后,由于不能被循环至膜表面,多数EGFR被分选至溶酶体发生降解,EGFR所介导的信号传导也被阻断,从而引起细胞凋亡。EE:早期内体;LE:晚期内体;Lysosome:溶酶体
3 研究展望
虽然已报道有多种技术可以用来靶向降解EGFR蛋白,但目前仍以基于蛋白降解系统的PROTAC技术的特异性最好。因为PROTAC技术在应用中可以采用针对肺癌中激酶区突变特异有效的小分子来进行蛋白降解药物的设计,而其他已报道的EGFR降解技术理论上均不能很好地区分野生型和激酶区突变的EGFR,这些技术对EGFR的选择性降解有待进一步研究,在后期应用中的毒副作用也有待评估。基于LYTAC技术开发的Ab-2通过识别EGFR胞外区发挥作用,机制上也不能够区分胞内部分是野生型还是EGFR激酶区突变体,在后期的应用中也难免会出现对正常细胞的毒副作用。同理,基于TRIB3和EGFR相互作用的SAH-JGZ4订书肽机制上也无法区分野生型EGFR和激酶区突变的EGFR,上述问题也难以避免。此外,这些不同的技术因具有各自的局限性,其成药性受到一定的影响。如利用LTR的LYTAC分子与抗体偶联,由于配体极性和分子量都很大,口服的可能性较小。
已有文献报道研究人员除了可以开发利用上述蛋白酶体系统和溶酶体系统的靶向蛋白降解技术之外,还开发了基于自噬系统的靶蛋白降解技术。自噬是细胞内另一种主要的蛋白降解机制。与蛋白酶体降解系统相比,自噬降解可以降解的底物蛋白更广泛。因此,借助自噬系统开发靶向蛋白降解剂,将开拓出更多可能性。目前,基于自噬系统开发的蛋白降解剂技术包括AUTAC(autophagy-targeting chimera)[39]和ATTEC(autophagosome-tethering compound)[40],并且已成功设计了针对MetAP2、FKBP12等多个靶点的AUTAC分子[39,41]。然而至今尚无基于自噬溶酶体降解系统开发的EGFR蛋白降解剂的报道,应用该技术开发的EGFR靶向降解药物的优劣仍需拭目以待。
由于EGFR蛋白在肺癌的发生发展中起着关键作用,特异性地靶向降解EGFR在肺癌的治疗中具有极大的发展潜力。此外,EGFR还与其他多种肿瘤的发生发展密切相关,因而开发不同于小分子抑制剂的EGFR靶向蛋白降解剂有着广阔的应用前景。目前,尚无能够有效降解EGFR C797S突变的蛋白降解剂被开发出来。蛋白降解技术虽很有潜力克服耐药现象,但是EGFR靶向蛋白降解剂的研究仍任重而道远。
